Missingness per Sequencing Project
Appendix A – Code to upload samples as they go + pipeline A
Appendix C – pipeline C Cram or Bam to FastQ
Appendix D- BAM to FASTQ processing (specific to the ADNI project)
Appendix F- Joint calling pipeline
Here, I summarize all the details involved in the processing and QC (Figure 1) of whole exome and whole genome sequence data for Alzheimers Disesease projects at the Cruchaga Lab at Washington University in St Louis. This report also provides brief summary of pipelines required to process sequencing data. Our latest datafreeze, Bloomfield, was completed in October 2021.
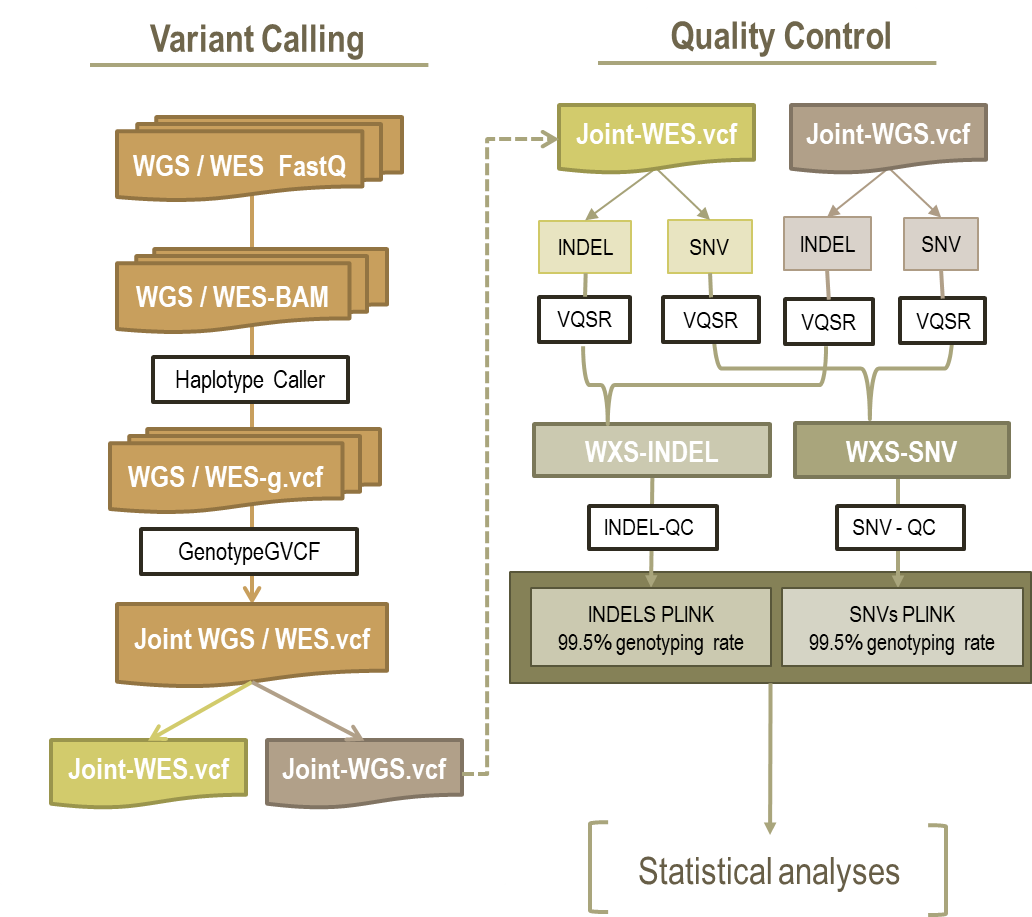
Figure 1. Summary of next generation sequence (NGS) data processing and quality control (QC) pipeline.
202110_bloomfield datafreeze consist of 9810 samples either in raw fastq, bam, or cram formats. To process these samples, several pipelines and Docker images were created implementing Bedtools (version 2.27.1), Samtools (version 1.9), Picard (version 2.20.8) and GATK (version 4.1.2.0). In summary, we have 48 projects (Table 1) that were processed using the same pipelines (Pipeline A, see Figure 1) in our computing platform at McDowell genome institute (MGI) at WashU.
All reference files required for variant calling are available on the local directory at the MGI server
/gscmnt/gc2645/wgs/Genome_Ref/GRCh38. Reference genome GRCh38 (GRCh38.p13; the thirteenth patch of the release) is available at MGI on /gscmnt/gc2645/wgs/Genome_Ref/GRCh38.
Files for dbSNP and high confidence SNPs and Indels calibration callsets from the 1K genome and Mills (Mills, Devine, Genome Research, 2011) public resources are also available at MGI on /gscmnt/gc2645/wgs/Genome_Ref/GRCh38/20190522_bundle.
Our pipeline was built to efficiently download/transfer raw files from external servers, immediately process them, and delete the raw and unnecessary files after successful completion of each variant calling step in the pipeline (See Appendix A)
In summary, here are the series of steps (in order) we followed to process these datasets; this pipeline assumes the starting file are fastq; for projects with different starting raw data, refer to “Alternative pipelines” section:
-
We started with creating a metafile (fields separated by commas) with the following contents for each sequencing project:
-
sample name (always possible to put final name, if different one provided try to change it)
-
DNA barcode (if available and provided by the lab or collaborator, alternatively we generated virtual barcodes)
-
sequencing project name (as specified in Table 1)
-
FULLSM (FULLSM was created as “${Sample_name}^${DNA_barcode}^${Project_name}”)
-
READ group, (which refers to a set of reads that are generated from a single run of a sequencing instrument was used to create a RGBASE as “${FULLSM}.${READ_group}”)
-
file name
-
file name extension ( identifier specified as a suffix to the name of either fastq, bam, cram – as inTable 1)
-
-
We performed BWA alignment of reads based on human genome build GRCh38 (GRCh38.p13; the thirteenth patch release for the GRCh38 reference assembly) using our docker image achalneupane/bwaref. Reference genome was downloaded from: https://console.cloud.google.com/storage/browser/genomics-public-data/resources/broad/hg38/v0
-
After performing the alignment of reads, we validated the bam output for any improper formatting, faulty alignments, incorrect flag values, etc., using Picard. We used our docker image achalneupane/validatesamref for this purpose. Any subsequent bam files produced by our pipelines were also “validated” using this tool.
-
We are also starting to include a docker image for VerifyBamID (achalneupane/verifybamid) that will calculate freemix values. These represent relative sample contamination.
-
We restricted our analysis to include only the exonic variants using Bedtools version 2.27.1 implemented in our docker image achalneupane/intersectbedref.
-
After performing the alignment for each RGBASE, we locate and tag the duplicate reads using Markduplicates tool in GATK version 4.1.2. Duplicates can originate from a single fragment of DNA either during library construction or also as optical duplicates. This tool differentiates the primary and duplicate reads using an algorithm that ranks reads by the sums of their base-quality scores. We then merge multiple bam files arising from each RGBASE into one single bam file.
-
We calculate depth of coverage using depthofcoverage tool in GATK to assess sequence coverage by a wide array of metrics, partitioned by sample, read group, or library. The docker image is available on dockerhub as achalneupane/depthofcoverage.
-
We generate a recalibration table for Base Quality Score Recalibration (BQSR) using BaseRecalibrator tool in GATK which was implemented in our docker image achalneupane/baserecalv2. Here, we performed a by-locus traversal operating only at sites that are in the known sites VCF. We used dbSNP and high confidence SNPs and Indels calibration callsets from the 1K genome and Mills (Mills, Devine, Genome Research, 2011) public resources as known sites of variation.
-
We used haplotype caller in GATK to call for germline SNPs and indels via local re-assembly of haplotypes implemented in our docker image achalneupane/haplocallerv2.
-
Finally, variant evaluation and refinement was performed using “VariantEval” tool in GATK calculating various quality control metrics. These metrics include the number of raw or filtered SNP counts; ratio of transition mutations to transversions; concordance of a particular sample's calls to a genotyping chip; number of s per sample, etc. This tool was implemented in our docker image achalneupane/variantevalref
In addition, we consistently used the same version of tools to process these datasets throughout our pipelines.
Any processed output files were immediately transferred to our local storage on Fenix. These files include, GATK report, coverage statistics report, recalibration tables, and variant calling (gVCF), and their index files. The aligned bam files after marking duplicates are also saved to Box to be used in future collaborations/in case of “easier” recalling needed.
-
For any projects that have raw reads in bam or cram format, we first unwrapped those samples into paired fastq files per read groups using RevertSam and SamToFastq tools in Picard version 2.20.8. This tool has been implemented in our docker image achalneupane/cram2fastq (see, appendix C).
-
Although the aforementioned pipeline was used to process BAM/CRAM samples, the pipeline to process BAM files from ADNI_WGS project was slightly different. The BAM files from this project did not have the RGIDs defined in the BAM header, so a new Docker image (achalneupane/bam2fqv2) was created that pulls the RGID from the read header itself and unwraps the BAM per RGID (see, appendix D). These files were relatively larger than the files from other projects. To speed up the process, we first unwrapped them on Compute 1 cluster and aligned the processed FASTQ (interleaved reads) per RGID on MGI.
-
We also downloaded samples for some projects from the dbGaP database in the SRA format. The interleaved sequence reads were extracted from the SRA files in FASTQ format that were processed using the pipeline E (see, Figure 1; Appendix E).
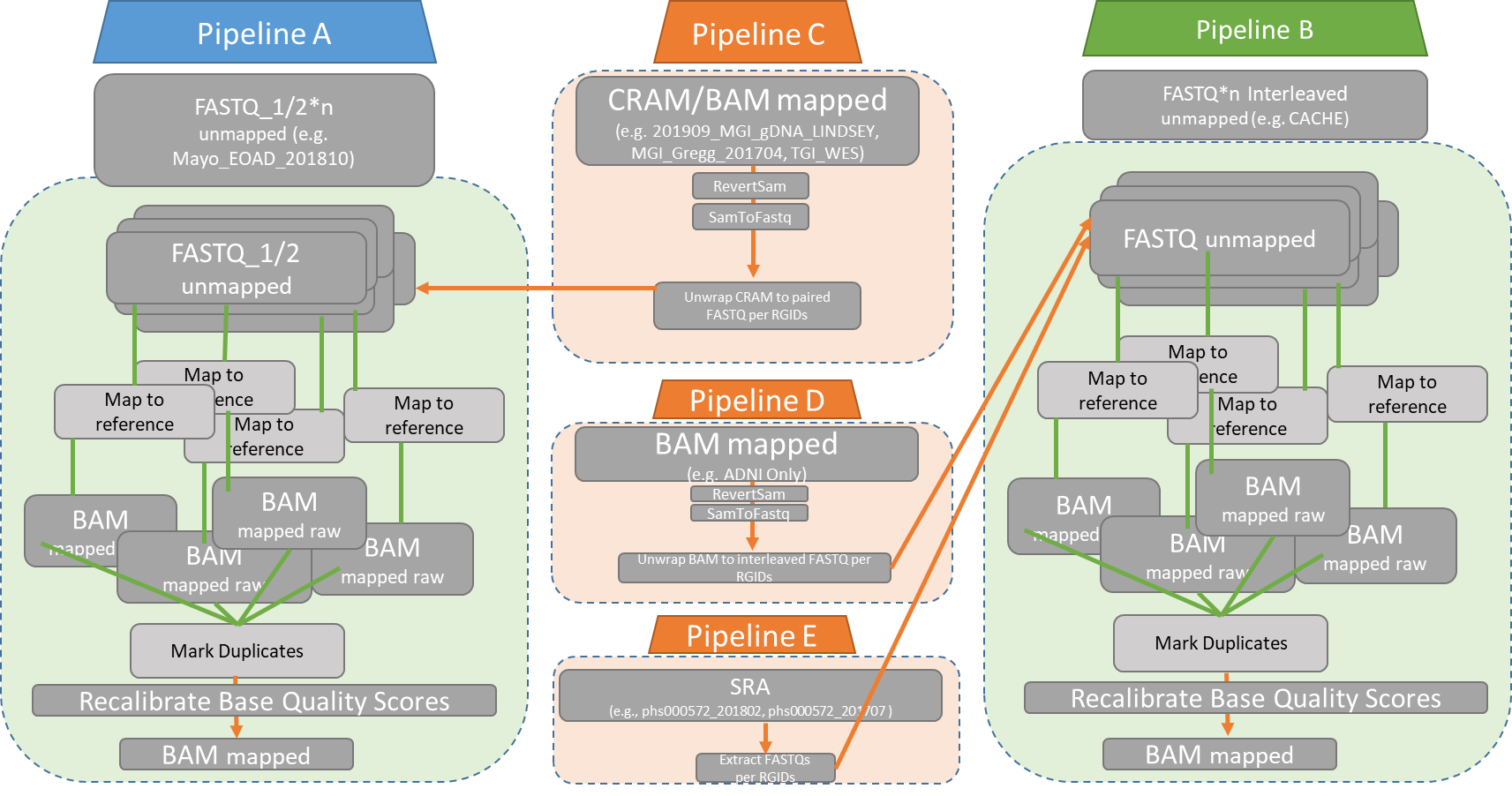
Figure 2 Summary of pipelines according to starting raw file format, and integration into the main pipeline. These pipeline are shown in Appendices A-E.
Table 1. Summary of sequencing projects included in the current datafreeze – Bloomfield. Details include nature of data (WXS being WES or WGS), nature of source data (fastq, bam or cram), number of samples per sequencing project and pileine (A to E) that samples for each particular project should follow.
| Seq project | WXS | source /raw data | # gVCFs | Pipeline |
|---|---|---|---|---|
| DIAN | WGS | r1.fq.gz | 12 | A |
| Macrogen_WGS | WGS | r1.fq.gz | 20 | A |
| MAPT_A152T | WGS | r1.fq.gz | 21 | A |
| LOAD_WES | WES | qual2sanger.r1.fq.gz (99%) / r1.fq.gz (1%) | 33 | A |
| Genentech_WES | WES | qual2sanger.r1.fq.gz / r1.fq.gz / qualsanger.r1.gz (1) | 91 | A |
| Genentech_WGS | WGS | qual2sanger.r1.fq.gz | 47 | A |
| 201812_MGI_DIANWGS_REDCLOUD | WGS | cram | 546 | C |
| 201909_MGI_gDNA_LINDSEY | WGS | cram | 637 | C |
| 202007_MGI_UPittKambohPiB_WGS_ELLINGWOOD | WGS | cram | 700 | C |
| phs000572_201508 (ADSP) | WES | sra | 117 | E |
| phs000572_201612 (ADSP) | WGS | sra | 190 | E |
| Broad_WGS | WGS | bam | 174 | C |
| TGI_WES | WES | bam | 298 | C |
| MGI_FASeEOAD_201605 | WES | bam | 423 | C |
| MGI_DIAN_201610 | WGS | bam | 4 | C |
| MGI_Imaging_201612 | WES | bam | 499 | C |
| Otogenetics_WES | WES | fq1/fq2 | 834 | A |
| phs000376 (familalPD) | WES | fq | 44 | B |
| phs000908 (Rare Variant PD) | WES | fq1/fq2 | 200 | A |
| phs000572_201707 (ADSP) | WES | sra | 94 | E |
| PPMI_WES | WES | fq | 591 | A |
| ADNI_WGS | WGS | bam | 809 | D |
| MGI_Gregg_201704 | WES | bam | 83 | C |
| phs000901(PD w/ CSF biomarker) | WES | fq | 57 | B |
| MGI_DIANEXR_201706 | WGS | bam | 19 | C |
| MGI_DIANEXR_201805 | WGS | bam | 3 | C |
| CACHE_WGS (Keoni) | WGS | fq | 215 | B |
| phs000572_201802(ADSP) | WES | sra | 1540 | E |
| MGI_DIANEXR_201902 | WGS | r1.fq.gz | 5 | A |
| Mayo_EOAD_201810 | WGS | fq1/fq2 | 227 | A |
| Mayo_Biobank-Control_201810 (PROCESS) | WGS | fq1/fq2 | 197 | A |
| Mayo_FTLD-TDP_201901 | WGS | fq | 11 | B |
| MGI_DIANEXR_201906 | WGS | r1.fq.gz | 2 | A |
| 201904_MGI_IDTexome_HURON | WES | cram | 34 | C |
| 201907_USUHS_gDNA_SHERMAN | WGS | fq1/fq2 | 45 | C |
| 202002_Mendelics_DIANEXR_WES_UNNAMED | WES | bam | 2 | C |
| 202004_AGRF_EOAD_WGS-WES_UNNAMED | WGS | 2 bam | 2 | C |
| 202103_ADSP_FUS-familial_WGS_UNNAMED | WGS | cram | 180 | C |
| 202004_USUHS_EOAD-WGS_gDNA_EOLUS | WGS | fq1/fq2 | 613 | A |
| 202104_ADSP_site27-sync-n303_WGS_UNNAMED | WGS | cram | 183 | C |
| 202008_MGI_DIAN_WGS-WES_UNNAMED | WGS | fq1/fq2 | 1 | A |
| 202009_MGI_DIAN_WGS-WES_UNNAMED | WGS | fq1/fq2 | 1 | A |
| 202011_MGI_DIAN_WGS-WES_UNNAMED | WGS | fq1/fq2 | 1 | A |
| 202103_MGI_DIAN_WGS_UNNAMED | WGS | fq1/fq2 | 1 | A |
| 202104_MGI_DIAN_WGS_UNNAMED | WGS | fq1/fq2 | 1 | A |
| 202104_MGI_DIAN_WGS-WES_UNNAMED | WES | fq1/fq2 | 1 | A |
| 202106_MGI_DIAN_WES_UNNAMED | WES | fq1/fq2 | 1 | A |
| 202106_MGI_DIAN_WGS_UNNAMED | WGS | fq1/fq2 | 1 | A |
| TOTAL | 9810 |
Joint calling of 9,810 samples was performed at the MGI. The Joint Calling is a two-step process.
First, we used GenomicsDBImport to combine GVCFs before performing joint
genotyping. We ran this step per chromosome.
GenomicsDBImport is used to import single-sample g.VCF and merge them into
GenomicsDB before joint genotyping. This step needs an input file listing all
the sample names and corresponding gVCF file names to include in a joint VCF.
The gVCF file names cannot have “^” and symlinks without “^” also did not work
with GenomicsDBImport, so temporary files were created replacing “^” with a
“.”.
We then performed joint genotyping of gVCFs on each chromosome using
GenotypeGVCFs tool in GATK which was implemented in docker image
broadinstitute/gatk:4.1.2.0.
See Appendix F for a full description of the pipeline.
The quality control (QC)
Here is a flowchart of the general QC done prior to Plink and the folder for performing all these steps is here:
${DIR}/01-Bloomfield-preQC/
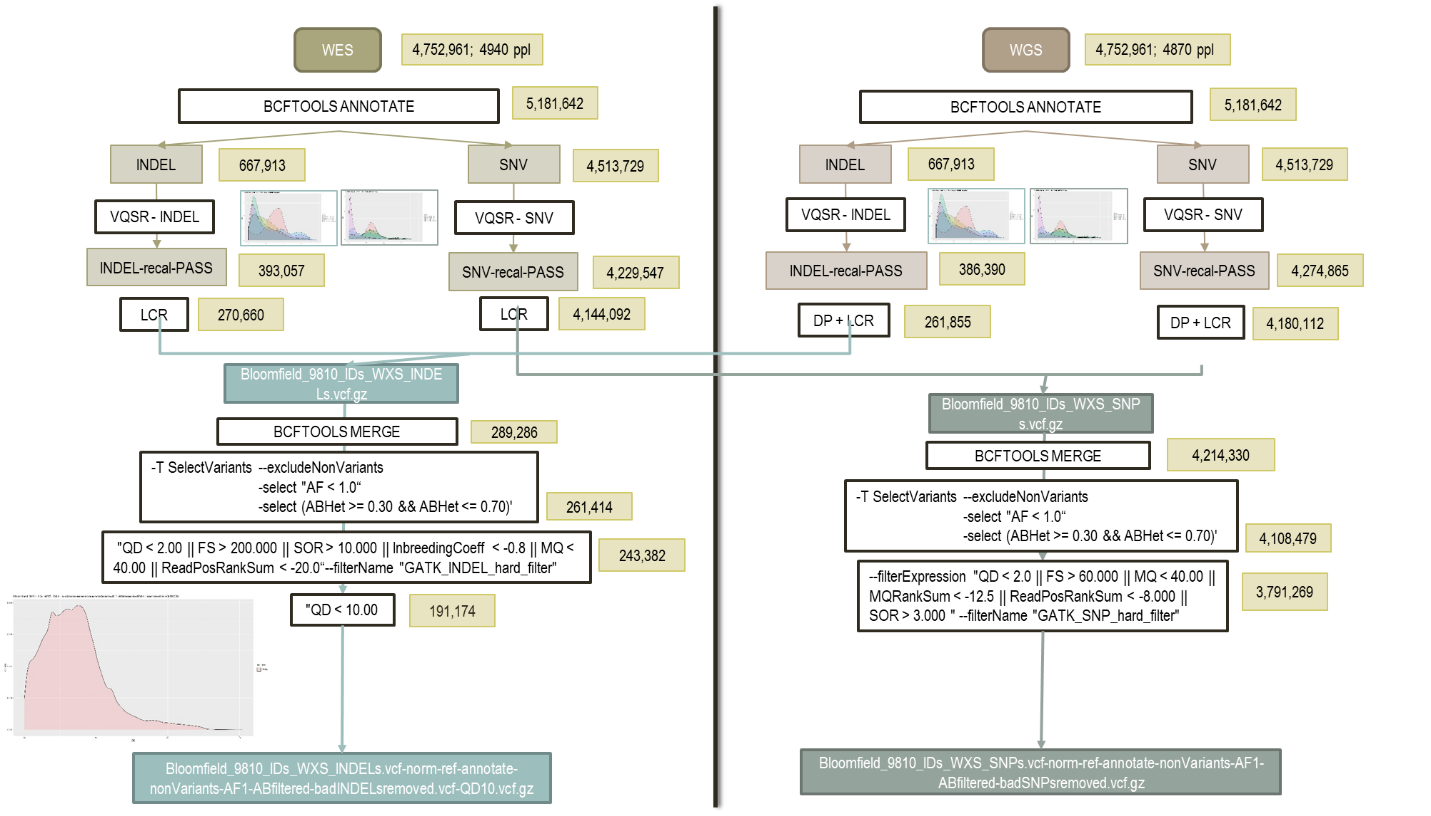
Figure 3. Shcematic view of the quality control steps performed through GATK.
Details of each of these steps are given next
The data is split into WES and WGS data. The variants are also split in each dataset as SNPs or INDELs as there are differences and requirements for filtering. Bcftools annotate is used to split VCF files from multi-allelic to bi-allelic, which is why variant numbers appear to increase at this step. Variant Quality Score Recalibration (VQSR) is done by ApplyVQSR in the GATK software. The algorithm in VQSR uses Gaussian mixture model that classifies variants based on how their annotation values cluster given a training set of high-confidence variants. The VQSR tool then uses this model to assign a new confidence score to each variant, called VQSLOD. This is a log-ratio of the variant’s probabilities belonging to the positive and negative model. For the Variant Recalibrator, we have had problems with some chromosomes failing due to not enough detected variants. To fix this, we had to decrease the max-gaussian option to the following values:
WGS SNVs: max-gaussian 6 for all except chrY which has a value of 1.
WGS INDELs: max-gaussian 2 for all except chr16 and chrY which has a value of 1.
WES SNVs: max-gaussian 6 for all chromosomes except 2 for chr13 and 1 for chrY.
WES INDELs: max-gaussian 2 for all chromosomes except for chr12, chr16, chr21, and chrY which have a value of 1.
All the analysis and files for the VQSR step is here:
${DIR}/01-Bloomfield-preQC/01-VQSR-ExAC-tsSNP99.6-tsINDEL95
The scripts for VQSR, 02a-VQSR-WGS-SNVs.sh and 02b-VQSR-WGS-INDELs.sh can be found here:
${DIR}/01-Bloomfield-preQC/
After VQSR, we remove variants in low-complexity regions of the DNA.
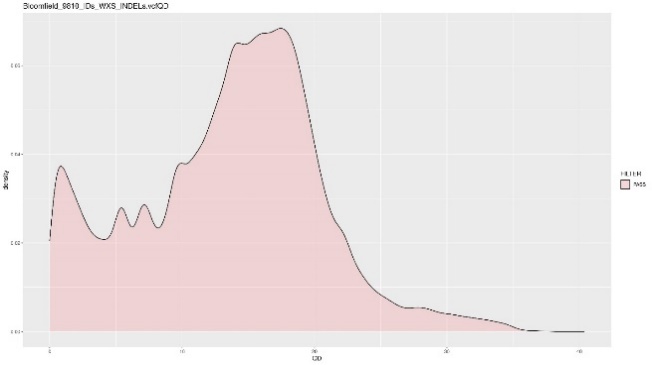
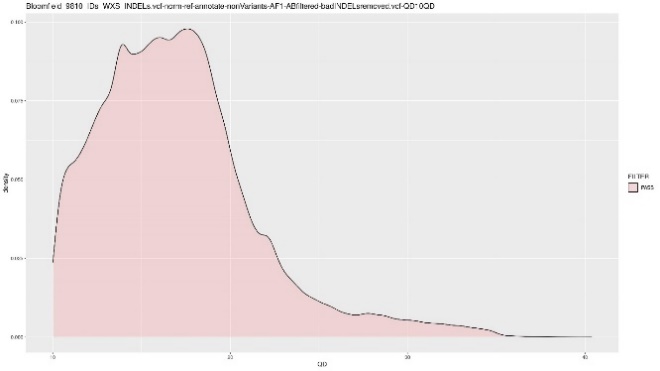
Before hard filtering, we combine WES and WGS data for both the SNPs and INDELs. Multi-allelic variants were separated into bi-allelic variants, and annotation and normalization was performed using Bcftools. Hard filtering includes getting rid of bad quality variants by removing non-variants and monomorphic variants. For the indels, we have to also manually filter by the qualbydepth score or the QD score. This is the variant confidence normalized by the unfiltered depth of variant samples. The plots below show the INDELs before and after removing all variants lower than a QD score of 10. Generally, we try to remove all of the smaller peaks on the lower left side of the original QD plot, so we get a distribution as “normal” as possible.
| QD plot – Number of variants = 243,382 | QD plot >= 10 – Number of Variants = 191,174 |
|---|---|
 |
 |
|
Hard filtering scripts for hard filtering including 03a_HQC_WXS_SNVs.sh and 03b-HQC_WXS_INDELs.sh are here:
/${DIR}/01-Bloomfield-preQC/
This is the general overview of the QC performed for PLINK. Next, there is a detail specification of each step and criteria for threshold when needed.
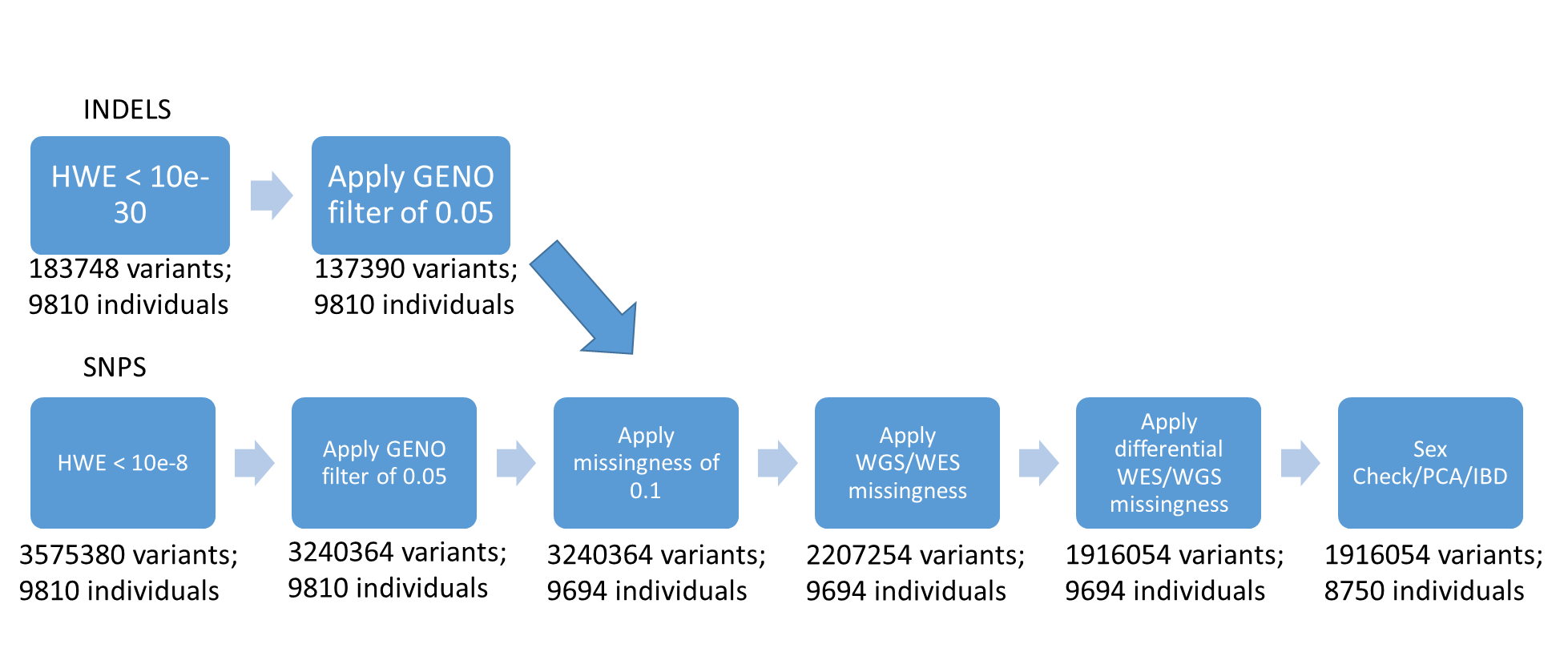
The QC on plink was done using these serial steps that will remove variants and samples that do not meet quality criteria:
-
Hwe filter of 10e-30 for INDELs and 10e-8 for SNPs
-
Geno filter of 0.05
-
Merge together SNPs and INDELs
-
Apply missingness per individual
-
Apply differential missingness – done for WXS and WGS, and for 15 different projects
-
Heterozigosity calculations
-
Sex-check
-
PCA
-
IBD
All files and logs for this can be found here:
/${DIR}/03-PLINK-QC-files
All variants were tested for Hardy-Weinberg equilibrium. This is common practice in genome wide association studies to detect variants that suffer from significant genotyping error.
BFILE_INDELS="Bloomfield_9810_INDELS"
plink1.9 --bfile \${BFILE_INDELS} --nonfounders --hwe 10e-30 include-nonctrl
\--keep-allele-order --autosome --make-bed --out \${BFILE_INDELS}-autosomes
BFILE_SNPS="Bloomfield_9810_SNPS"
plink1.9 --bfile \${BFILE_SNPS} --nonfounders --hwe 10e-8 include-nonctrl
\--keep-allele-order --autosome --make-bed --out \${BFILE_SNPS}-autosomes
plink1.9 --bfile \${BFILE_INDELS} --chr X,Y,XY --make-bed --keep-allele-order
\--out \${BFILE_INDELS}-X-Y-XY
plink1.9 --bfile \${BFILE_SNPS} --chr X,Y,XY --make-bed --keep-allele-order
\--out \${BFILE_SNPS}-X-Y-XY
\# Merge autosomes and chr X, Y, XY
plink1.9 --bfile \${BFILE_INDELS}-autosomes --bmerge \${BFILE_INDELS}-X-Y-XY.bed
\${BFILE_INDELS}-X-Y-XY.bim \${BFILE_INDELS}-X-Y-XY.fam --keep-allele-order
\--merge-mode 4 --indiv-sort 0 --out \${BFILE_INDELS}-hwe
plink1.9 --bfile \${BFILE_SNPS}-autosomes --bmerge \${BFILE_SNPS}-X-Y-XY.bed
\${BFILE_SNPS}-X-Y-XY.bim \${BFILE_SNPS}-X-Y-XY.fam --keep-allele-order
\--merge-mode 4 --indiv-sort 0 --out \${BFILE_SNPS}-hwe
Percent of variants removed from applying the HWE flag at various p-values (Table 2, Table 3); based on these values, and what is currently used in the literature by similar studies, we decided to
Table 2. number of variants removed in the SNPs and INDELs datasets according to differnet p-value thresholds.
| P.Value | SNPs | INDELs |
|---|---|---|
| 0.05 | 9.099% | 16.254% |
| 10e-6 | 2.676% | 6.738% |
| 10e-8 | 1.991% | 5.425% |
| 10e-30 | 0.407% | 1.7605 |
| Bonferroni threshold | 1.759% (1.37e-8) | 5.664% (2.67e-7) |
Table 3. DeFinetti plots for HWE values for SNPs and INDELS, pre and post filtering
| SNPs | Pre-HWE Pre hwe filter – 10e-6 | Post – HWE Filtered using 10e-8 |
|---|---|---|
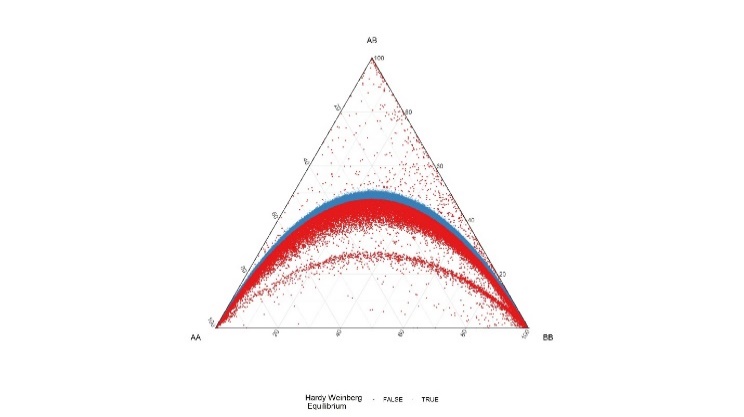 |
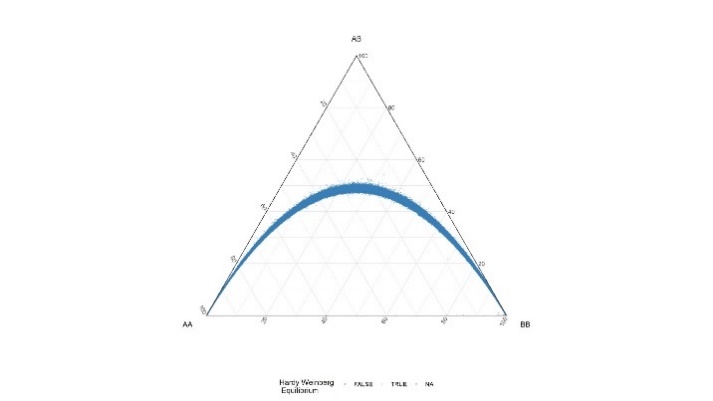 |
| INDELs | Pre-HWE mark for 10e-6 | Post – HWE Filtered using 10e-30 |
|---|---|---|
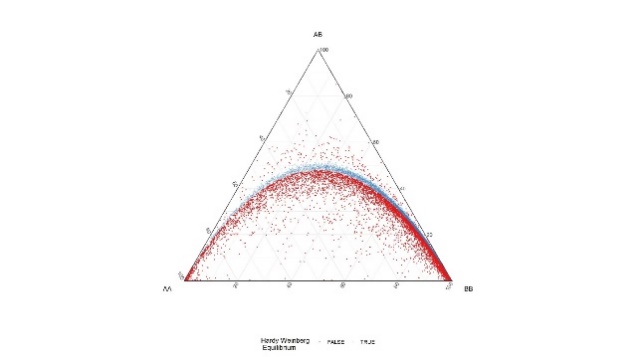 |
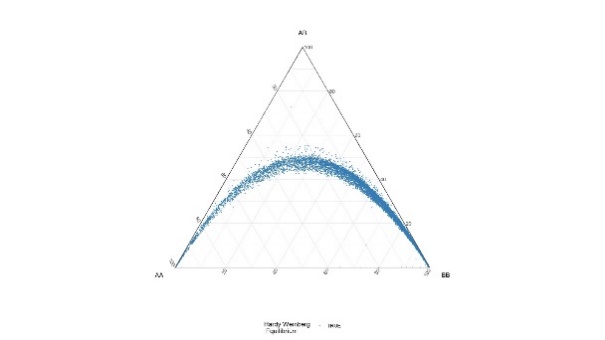 |
The genotyping rate or –geno filter gets rid of all variants with missing call rates over 0.05, or whatever value you prefer. In our dataset we use a value of 0.05 for SNPs and INDELs.
GENO1="0.05"
plink1.9 --bfile \${BFILE_INDELS}-hwe --geno \${GENO1} --make-bed
\--keep-allele-order --allow-no-sex --out \${BFILE_INDELS}-hwe-geno\${GENO1}
plink1.9 --bfile \${BFILE_SNPS}-hwe --geno \${GENO1} --make-bed
\--keep-allele-order --allow-no-sex --out \${BFILE_SNPS}-hwe-geno\${GENO1}
This filter removed 472,406 SNP variants and 46,358 INDEL variants. The resulting genotyping rate is 0.9080 for INDELs and 0.9885 for SNPs.
The –mind filter gets rid of samples in your dataset with a missing call rate above a certain value. This is similar to the –geno filter but instead of removing variants we are removing samples from further analysis.
plink1.9 --bfile \${BFILE}-hwe-geno\${GENO1} --mind 0.1 --make-bed
\--keep-allele-order --out \${BFILE}-hwe-geno\${GENO1}-mind0.1
There were 116 samples removed by the –mind filter. Table 4 The samples below show the number of individuals removed and from which project they belong. Samples from phs000901 were known to have extremely low coverage with 0.005% bases above 10x coverage.
Table 4. Relationship of samples per project that failed missingness 0.1 filter.
| Seq Project | # samples with missingness > 0.1 |
|---|---|
| phs000901 | 57 |
| CACHE_WGS | 4 |
| PPMI_WES | 33 |
| MGI_Gregg_201704 | 2 |
| Otogenetics_WES | 2 |
| MGI_Imaging_201612 | 3 |
| phs000908 | 12 |
| 201909_MGI_gDNA_LINDSEY | 1 |
This dataset contains a mixture of WES and WGS data Table 5; those two types of sequence data are generated through differnet procudures which can give place to variants that exclusively found in the WES or in the WGS dataset. To avoide this batch effect, we perform differential missingness using WES or WGS as “phenotype” with the plink command –test-missing. The METADATA file contains a WXS column where 1 represents WGS sequencing type and 2 represents WES.
BFILE="Bloomfield_9810-hwe-geno0.05-mind0.1"
METADATA="${DIR}/01-Bloomfield-preQC/Bloomfield_9810_metadata.txt"
plink1.9 --bfile ../02-intermediate-filtering-files/\${BFILE} --pheno
\${METADATA} --pheno-name WXS --test-missing --allow-no-sex --out
\${BFILE}-missing-WXS
Variants with differential missingness between WES and WGS data sets, and a Hardy-Weinberg equilibrium of p < 1e-8, were removed from analysis.
FILE2="\${BFILE}-missing-WXS.missing"
plink1.9 --bfile ../02-intermediate-filtering-files/\${BFILE} --exclude
\${FILE2%.\*}.list --keep-allele-order --make-bed --out \${BFILE}-WXSm
A total of 1,033,110 variants were removed and the final genotyping rate is
0.99939.
Table 5. Proportion of WES and WGS in the dataset
| WXS | # | % |
|---|---|---|
| WES | 4940 | 50.4% |
| WGS | 4870 | 49.6% |
| TOTAL | 9810 |
Similar to the previous step, this dataset contains samples coming from multiple sequencing rounds and centers which can also be a source of batch effects; to minimize batch effect, we also perform differential missingness using sequencing project as the “phenotype” with the plink command –test-missing. We perform the differential missingness step for each project that contributes with more than 2% of total samples.
Table 6. Relationship fo sequencing projects, number of smaplesper project and % of samples contributing to the entire datafreeze.
| Sequencing project | N of samples | % of samples | # variants identified by differential missigness |
|---|---|---|---|
| 201812_MGI_DIANWGS_REDCLOUD | 546 | 5.56575 | |
| 201904_MGI_IDTexome_HURON | 34 | 0.346585 | |
| 201907_USUHS_gDNA_SHERMAN | 45 | 0.458716 | |
| 201909_MGI_gDNA_LINDSEY | 637 | 6.49337 | |
| 202002_Mendelics_DIANEXR_WES_UNNAMED | 2 | 0.0203874 | |
| 202004_AGRF_EOAD_WGS-WES_UNNAMED | 2 | 0.0203874 | |
| 202004_USUHS_EOAD-WGS_gDNA_EOLUS | 613 | 6.24873 | |
| 202007_MGI_UPittKambohPiB_WGS_ELLINGWOOD | 700 | 7.13558 | |
| 202008_MGI_DIAN_WGS-WES_UNNAMED | 1 | 0.0101937 | |
| 202009_MGI_DIAN_WGS-WES_UNNAMED | 1 | 0.0101937 | |
| 202011_MGI_DIAN_WGS-WES_UNNAMED | 1 | 0.0101937 | |
| 202103_ADSP_FUS-familial_WGS_UNNAMED | 180 | 1.83486 | |
| 202103_MGI_DIAN_WGS_UNNAMED | 1 | 0.0101937 | |
| 202104_ADSP_site27-sync-n303_WGS_UNNAMED | 183 | 1.86544 | |
| 202104_MGI_DIAN_WGS_UNNAMED | 1 | 0.0101937 | |
| 202104_MGI_DIAN_WGS-WES_UNNAMED | 1 | 0.0101937 | |
| 202106_MGI_DIAN_WES_UNNAMED | 1 | 0.0101937 | |
| 202106_MGI_DIAN_WGS_UNNAMED | 1 | 0.0101937 | |
| ADNI_WGS | 809 | 8.24669 | |
| Broad_WGS | 174 | 1.7737 | |
| CACHE_WGS | 215 | 2.19164 | |
| DIAN | 12 | 0.122324 | |
| Genentech_WES | 91 | 0.927625 | |
| Genentech_WGS | 47 | 0.479103 | |
| LOAD_WES | 33 | 0.336391 | |
| Macrogen_WGS | 20 | 0.203874 | |
| MAPT_A152T | 21 | 0.214067 | |
| Mayo_Biobank-Control_201810 | 197 | 2.00815 | |
| Mayo_EOAD_201810 | 227 | 2.31397 | |
| Mayo_FTLD-TDP_201901 | 11 | 0.11213 | |
| MGI_DIAN_201610 | 4 | 0.0407747 | |
| MGI_DIANEXR_201706 | 19 | 0.19368 | |
| MGI_DIANEXR_201805 | 3 | 0.030581 | |
| MGI_DIANEXR_201902 | 5 | 0.0509684 | |
| MGI_DIANEXR_201906 | 2 | 0.0203874 | |
| MGI_FASeEOAD_201605 | 423 | 4.31193 | |
| MGI_Gregg_201704 | 83 | 0.846075 | |
| MGI_Imaging_201612 | 499 | 5.08665 | |
| Otogenetics_WES | 834 | 8.50153 | |
| phs000376 | 44 | 0.448522 | |
| phs000572_201508 | 117 | 1.19266 | |
| phs000572_201612 | 190 | 1.9368 | |
| phs000572_201707 | 94 | 0.958206 | |
| phs000572_201802 | 1540 | 15.6983 | |
| phs000901 | 57 | 0.58104 | |
| phs000908 | 200 | 2.03874 | |
| PPMI_WES | 591 | 6.02446 | |
| TGI_WES | 298 | 3.03772 |
Based on the table above, the sequencing projects that contribute with more than 2% of total samples are:
phs000908, Mayo_Biobank-Control_201810, CACHE_WGS, Mayo_EOAD_201810, TGI_WES, MGI_FASeEOAD_201605, MGI_Imaging_201612, 201812_MGI_DIANWGS_REDCLOUD, PPMI_WES, 202004_USUHS_EOAD-WGS_gDNA_EOLUS, 201909_MGI_gDNA_LINDSEY, 202007_MGI_UPittKambohPiB_WGS_ELLINGWOOD, ADNI_WGS, Otogenetics_WES, phs000572_201802
To perform differential missingness we add as many dummy variables as sequencing projects with the values “1” or “2” representing whether each sample has been sequenced (1) or not (2) under that particular sequencing project. Next, --test-missing is run as many times as sequencing projects identified.
plink1.9 --bfile \${BFILE} --pheno \${PHENOSCOPE} --pheno-name
Mayo_Biobank-Control_201810 --test-missing --allow-no-sex --out
\${BFILE}-scope_missing15
Then, all the output lists are collapsed into a single one and variants with a p-value p<1e-8 are removed from the dataset.
FILE="\${BFILE}-scope_missing15.missing"
sed -i 's/ \\+/\\t/g' \${FILE}
sed -i 's/\^\\t//g' \${FILE}
awk '{if (\$5\<=1.00e-08) print \$0}' \${FILE} \| cut -f2 \>\>
\${BFILE}.scope-missing.list
\#\# Put "BAD" variants in a file and remove from dataset - Part to do manually
awk '{if (\$5\<=1.00e-08) print \$0}' \${FILE2} \| cut -f2 \> \${FILE2%.\*}.list
\#\# NOW extract these BAD variants
plink1.9 --bfile \${BFILE} --exclude \${FILE2%.\*}.list --keep-allele-order
\--make-bed --out \${BFILE}-WXSm
There were 278,143 unique variants to remove from missingness per sequencing project.
All commands and files were run here:
${DIR}/01-Bloomfield-preQC/03-PLINK-QC-files
#Fenix
BFILE="Bloomfield_9810-hwe-geno0.05-mind0.1-WXSm"
plink1.9 --bfile \${BFILE} --het --out \${BFILE}_QC_het
sed -i "s/[[:space:]]\\+/\\t/g" \${BFILE}_QC_het.het
sed -i 's/\^\\t//g' \${BFILE}_QC_het.het
#R
het \<- read.table("Bloomfield_9810-hwe-geno0.05-mind0.1-WXSm_QC_het.het",
head=TRUE)
pdf("heterozygosity.pdf")
het\$HET_RATE = (het\$"N.NM." - het\$"O.HOM.")/het\$"N.NM."
hist(het\$HET_RATE, xlab="Heterozygosity Rate", ylab="Frequency", main=
"Heterozygosity Rate")
dev.off()
\#\#\#\#\# Find samples whoes het rate varies 3sd from the mean. using SNPs that
are not in LD (R_check)
\`het \<- read.table("Bloomfield_9810-hwe-geno0.05-mind0.1-WXSm_QC_het.het",
head=TRUE)
het\$HET_RATE = (het\$"N.NM." - het\$"O.HOM.")/het\$"N.NM."
het_fail = subset(het, (het\$HET_RATE \<
mean(het\$HET_RATE)-3\*sd(het\$HET_RATE)) \| (het\$HET_RATE \>
mean(het\$HET_RATE)+3\*sd(het\$HET_RATE)));
het_fail\$HET_DST = (het_fail\$HET_RATE-mean(het\$HET_RATE))/sd(het\$HET_RATE);
write.table(het_fail, "fail-het-qc.txt", row.names=FALSE, quote=F, sep="\\t")
Here is a plot of the heterozygosity rates for each sample:
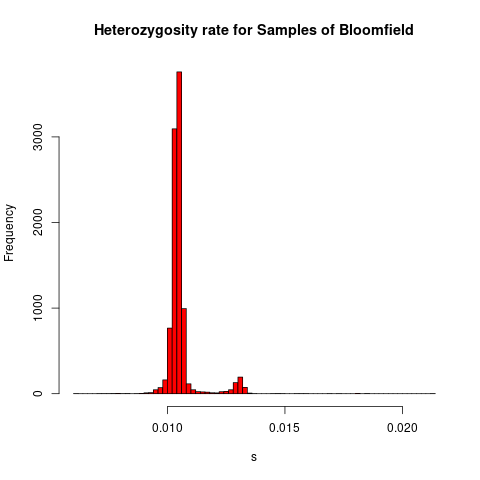
All samples that were more than 3 standard deviations from the mean were added to fail-het-qc.txt.
I ran the sex check according to the code below.
BFILE="Bloomfield_9810-hwe-geno0.05-mind0.1"
plink --bfile \${BFILE} --update-sex Bloomfield-gVCFID-SEX.csv --make-bed --out
\${BFILE}_with_sex
plink1.9 --bfile \${BFILE}_with_sex --check-sex --out \${BFILE}_sex
I filtered only those individuals with 'PROBLEM' listed in Bloomfield_9810-hwe-geno0.05-mind0.1_sex.sexcheck. Then, I filtered again based on there being a 1 or 2 in the PEDSEX and SNPSEX columns so that we take individuals that have information. The final list of individuals with different reported sexes are here:
${DIR}/01-Bloomfield-preQC/02-intermediate-filtering-files/Bloomfield_sex_check_problems2.txt.
There are a total of 25 individuals.
We created a PCA plot to determine the ethnic makeup of our dataset. Here is the code:
BFILE="Bloomfield_9810-hwe-geno0.05-mind0.1_with_STATUS"
plink1.9 --bfile \${BFILE} --allow-no-sex --cluster --geno 0.01 --genome --hwe
0.001 --ld-window-r2 0.2 --maf 0.01 --mds-plot 4 --min 0.2 --nonfounders --pca
header --out \${BFILE}-PCAS
#R
library(ggplot2)
PCAs\<-
read.table("Bloomfield_9810-hwe-geno0.05-mind0.1_with_STATUS-PCAS.eigenvec",
header=T)
ggplot(PCAs, aes(x=PC1, y=PC2)) + geom_point() + xlab("PC1") + ylab("PC2") +
ggtitle("Bloomfield-9694-PCA")
ggsave("Bloomfield-PCA.jpg", plot = last_plot(), device = NULL, scale = 1, width
= 16, height = 9, dpi = 300, limitsize = TRUE)
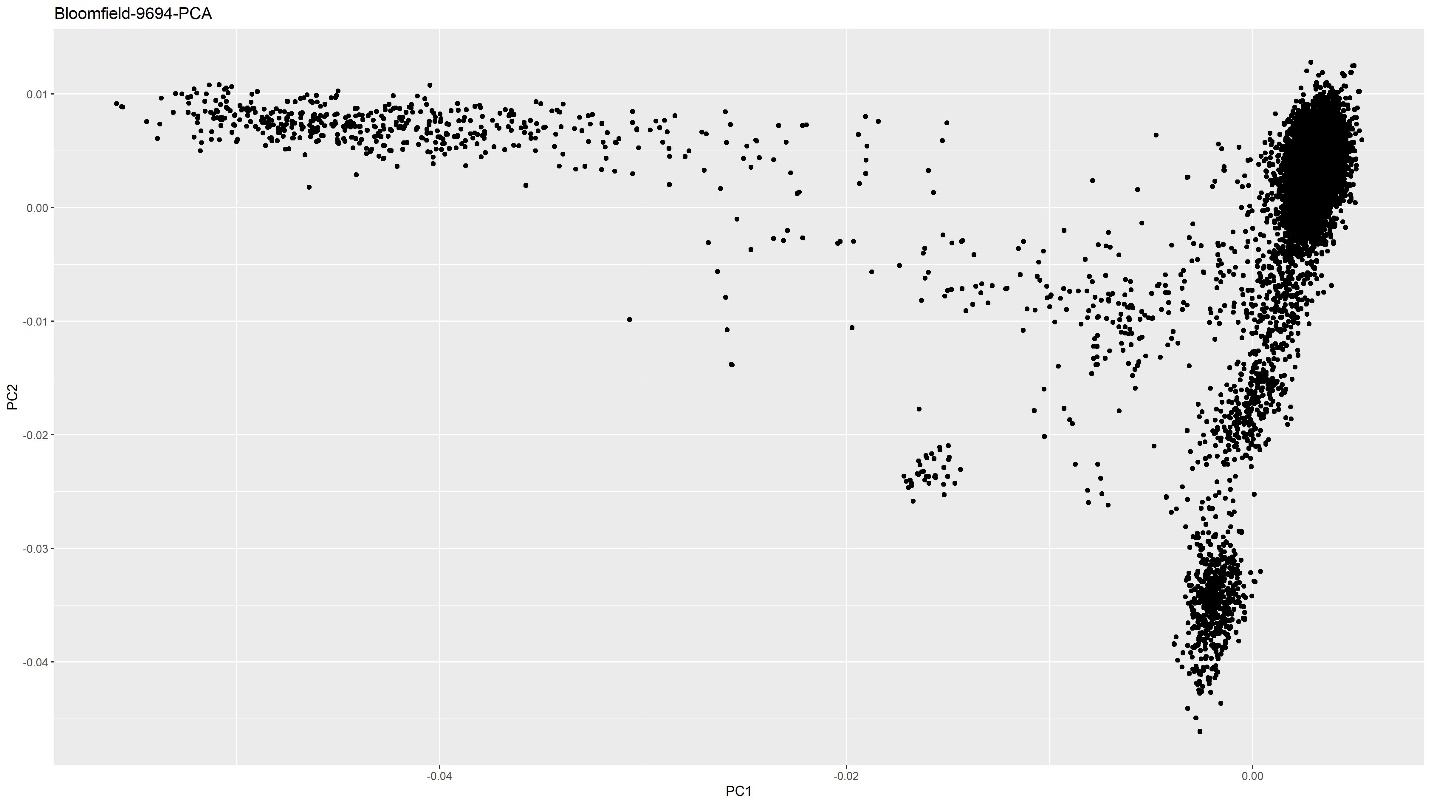
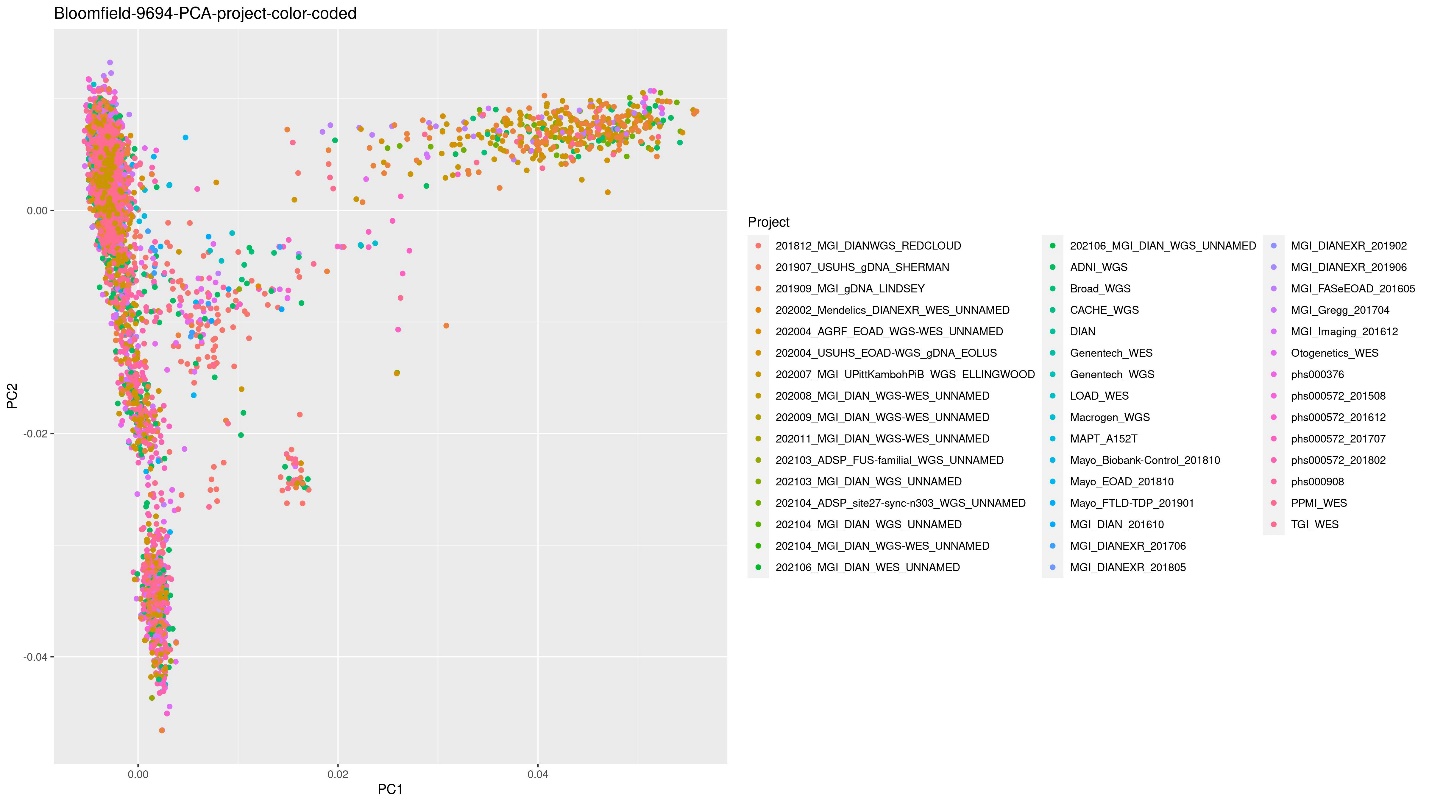
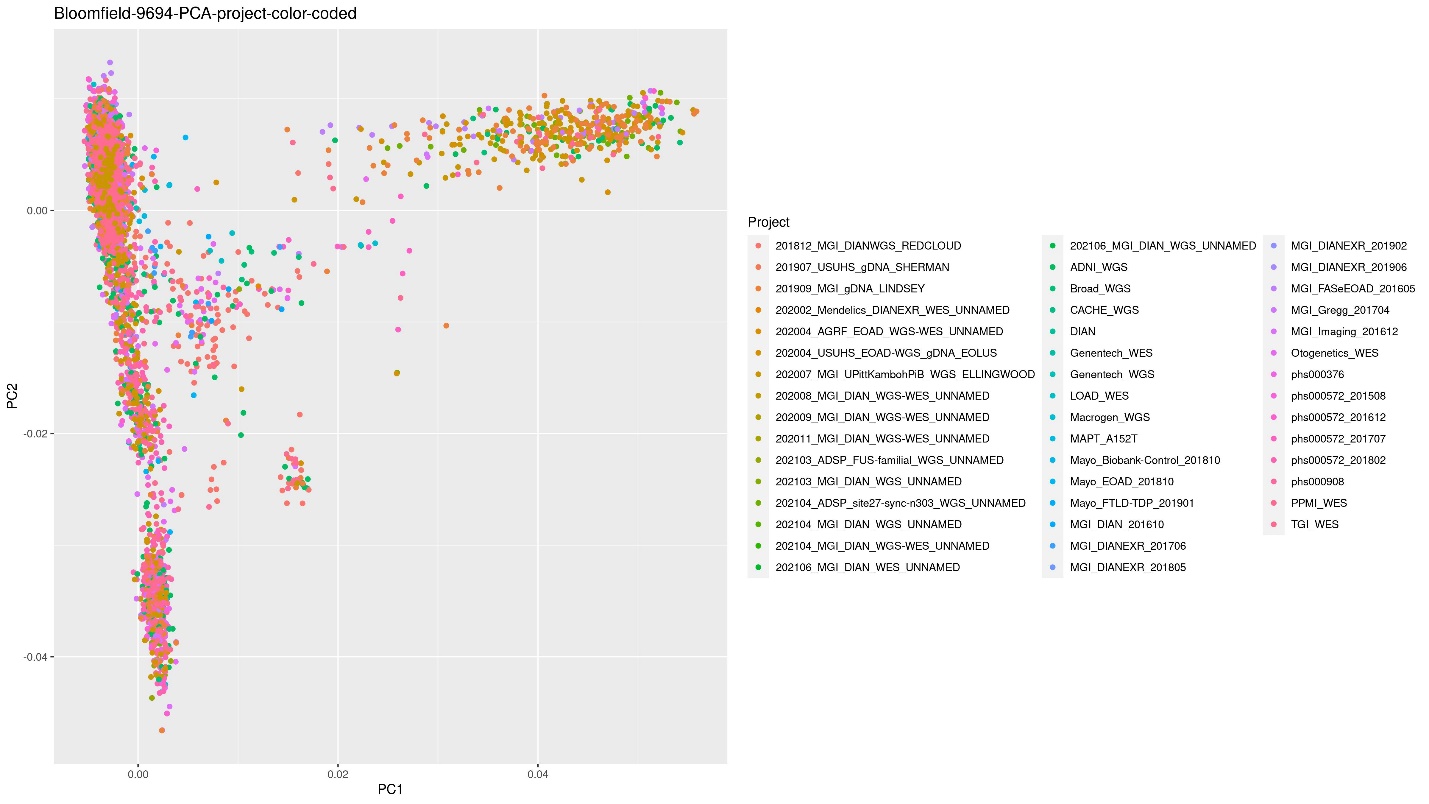
Similarly, the PCA color coded by project was created by adding the column that holds the project to the data variable as the Color:
ggplot(PCA2, aes(x=PC1, y=PC2, color=Project)) + geom_point() + xlab("PC1") +
ylab("PC2") + ggtitle("Bloomfield-9694-PCA-project-color-coded")
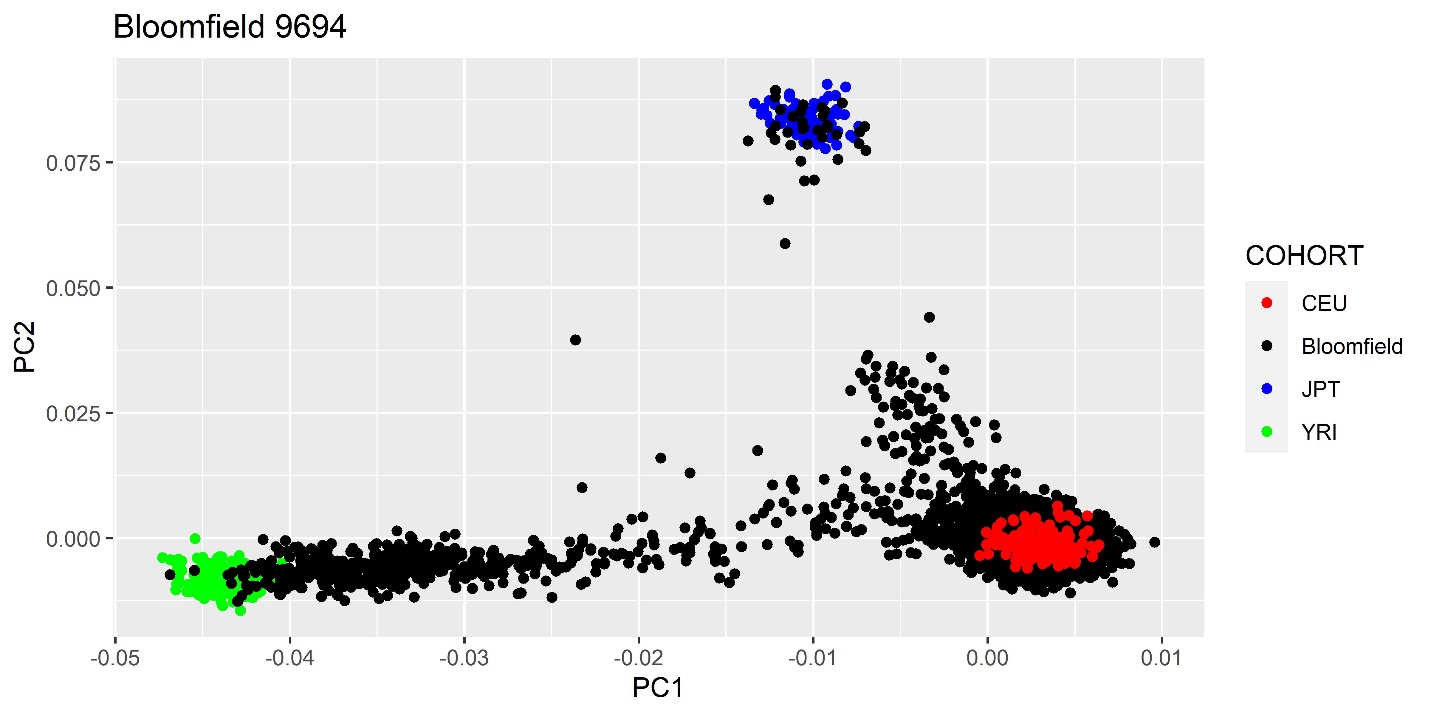
In order to view this PCA plot with known ethnic groups via Hapmap, I used the code below in R:
# # I used this code to anchor HAPMAP samples for plotting PCA
PCA \<-
read.table("Bloomfield_9810-hwe-geno0.05-mind0.1_with_STATUS-HAPMAP-MERGED3-for_PCA_no_mind.eigenvec",
header =T, stringsAsFactors=FALSE)
HAPMAP.ethnicty \<- read.table("relationships_w_pops_121708.txt", header = T )
head(HAPMAP.ethnicty)
PCA\$COHORT \<- "Bloomfield"
PCA\$COHORT \<- HAPMAP.ethnicty\$population[match(PCA\$IID,
HAPMAP.ethnicty\$IID)]
PCA \<- PCA[c(1:4,23)]
PCA\$COHORT \<- as.character(PCA\$COHORT)
PCA\$COHORT[is.na(PCA\$COHORT)] \<- "Bloomfield"
write.table(PCA,
"Bloomfield_WXS_SNPS_INDELS-hwe-geno0.05-mind0.1-HAPMAP-MERGED3-for_PCA.eigenvec-PC1-PC2-COHORT.txt",
sep ="\\t", col.names = T, quote = F)
\#\#\# PLOT PCAs in R
library(ggplot2)
PCAs\<-
read.table("Bloomfield_WXS_SNPS_INDELS-hwe-geno0.05-mind0.1-HAPMAP-MERGED3-for_PCA.eigenvec-PC1-PC2-COHORT.txt",
header=T)
\#\#plotting:
target \<- c("CEU", "Bloomfield", "JPT", "YRI")
PCAs\$COHORT \<- factor(PCAs\$COHORT, levels = target)
PCAs \<- PCAs[order(-as.numeric(factor(PCAs\$COHORT))),]
ggplot(PCAs, aes(x=PC1, y=PC2, color=COHORT)) + geom_point() + xlab("PC1") +
ylab("PC2") + ggtitle("Bloomfield 9694") +
scale_color_manual(values = c('red','black','blue', "green"))
ggsave("Bloomfield_9694_Hapmap_PCA.jpg", plot = last_plot(), device = NULL,
scale = 1, width = 8, height = 4, dpi = 600, limitsize = TRUE)
It is also important to note that you should take the mind flag out of the hapmap plink command. I caused the Hapmap data to de-anchor from the Bloomfield data. Here is the PCA plot with Hapmap:
Identity by Descent is a way to study the relatedness of samples in your dataset. Below is the code that I used to create the plot below for this dataset.
BFILE="Bloomfield_9810-hwe-geno0.05-mind0.1_with_STATUS"
plink1.9 --bfile \${BFILE} --geno 0.01 --genome --hwe 0.01 --ld-window-r2 0.2
\--maf 0.15 --out \${BFILE}-IBD
library(ggplot2)
IBD\<-read.table("Bloomfield_9810-hwe-geno0.05-mind0.1_with_STATUS-IBD.genome",
head=T)
ggplot(IBD, aes(x=Z0, y=Z1))+ geom_point() + ggtitle("Bloomfield-9694-IBD")
ggsave("Bloomfield-IBD.jpg", plot = last_plot(), device = NULL, scale = 1, width
= 16, height = 9, dpi = 300, limitsize = TRUE)
# Run the pipeline starting with the $START line number in the WORKLIST; WORKLIST is the list of all FULLSMs to be processed for gVCF. $number is the Nth sample being processed in the $WORKLIST.
export BASE="/gscmnt/gc2645/wgs"; \\
export WORKDIR="\${BASE}/tmp"; \\
export THREADS=16; \\
export BWA_PIPE_SORT=1; \\
export TIMING=1; \\
LOOKUP_COL_SM=1; \\
LOOKUP_COL_DNA=2; \\
LOOKUP_COL_PR=3; \\
LOOKUP_COL_RGBASE=4; \\
LOOKUP_COL_FQ1EXT=5; \\
LOOKUP_COL_FQ2EXT=6; \\
export RUN_TYPE="paddedexome"; \\
for FULLSM in \$(sed -n "\${START},\${END}p" "\${WORKLIST}"); do \\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo "Doing sample number\*\*\*\*\*\*\*\*\*\*: " \$Snumber; \\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
((Snumber=\${Snumber}+1)); \\
SM="\$(echo "\${FULLSM}" \| cut -d\^ -f1)"; \\
IFS=\$'\\n' export DNA=(\$(awk -F, "\\\$\${LOOKUP_COL_SM} == \\"\${SM}\\""
"\${LOOKUP}" \| cut -d, -f\${LOOKUP_COL_DNA} \| sort -u)); \\
if [ \${\#DNA[@]} -gt 1 ]; then echo "Warning, \\\${DNA} not unique for \${SM}
(n=\${\#DNA[@]}: \${DNA[@]})"; fi; \\
DNA="\$(echo "\${FULLSM}" \| cut -d\^ -f2)"; \\
PR="\$(echo "\${FULLSM}" \| cut -d\^ -f3)"; \\
export OUT_DIR="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}"; \\
echo -e "00 - Starting jobs per sample FULLSM \${FULLSM}"; \\
for RGBASE in \$(grep "\${SM},\${DNA},\${PR}" "\${LOOKUP}" \| cut -d,
\-f\${LOOKUP_COL_RGBASE}); do \\
SM="\$(echo "\${FULLSM}" \| cut -d\^ -f1)"; \\
DNA="\$(echo "\${FULLSM}" \| cut -d\^ -f2)"; \\
PR="\$(echo "\${FULLSM}" \| cut -d\^ -f3)"; \\
FBASE="/40/AD/AD_Seq_Data/01.-RawData/201907_USUHS_gDNA_SHERMAN/01.-RawData"; \\
\# folder where RG files are located \\
RBASE="/home/achal/01.-RawData/201907_USUHS_gDNA_SHERMAN/201907_USUHS_gDNA_SHERMAN";
\\
FQ1EXT=(\$(awk -F, "\\\$\${LOOKUP_COL_RGBASE} == \\"\${RGBASE}\\"" "\${LOOKUP}"
\| cut -d, -f\${LOOKUP_COL_FQ1EXT})); \\
FQ2EXT=(\$(awk -F, "\\\$\${LOOKUP_COL_RGBASE} == \\"\${RGBASE}\\"" "\${LOOKUP}"
\| cut -d, -f\${LOOKUP_COL_FQ2EXT})); \\
RGFILE="\${RBASE}/\${RGBASE}.rgfile"; \\
FQ1="\${FBASE}/\${RGBASE}\${FQ1EXT}"; \\
FQ2="\${FBASE}/\${RGBASE}\${FQ2EXT}"; \\
DEST="\${BASE}/WXS_Aquilla/01-RAW"; \\
echo -e "00a - Uploading FASTQ and rgfiles for sample \${FULLSM} and RGBASE
\${RGBASE}\\nFQ1:\${FQ1}\\nFQ2:\${FQ2}\\nRGFILE:\${RGFILE}\\nDNA:\${DNA}"; \\
mkdir \${DEST}/\${PR}/\${FULLSM}; \\
\#\#\# rsync with copy referent files as we are copying symlinks for this PPMI
data from fenix
rsync -avh -L \${USER}@fenix.psych.wucon.wustl.edu:\${FQ1}
\${DEST}/\${PR}/\${FULLSM}/; \\
rsync -avh -L \${USER}@fenix.psych.wucon.wustl.edu:\${FQ2}
\${DEST}/\${PR}/\${FULLSM}/; \\
rsync -avh \${USER}@fenix.psych.wucon.wustl.edu:\${RGFILE}
\${DEST}/\${PR}/\${FULLSM}/; \\
export RGFILE="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.rgfile";
\\
export FULLSM_RGID="\${RGBASE}"; \\
unset BAMFILE; \\
echo -e "01 - Starting bwa per sample \${FULLSM} and RGBASE \${RGBASE}"; \\
export RGFILE="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.rgfile";
\\
export FQ1="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}\${FQ1EXT}";
\\
export FQ2="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}\${FQ2EXT}";
\\
export REF="\${BASE}/Genome_Ref/GRCh38/Homo_sapiens_assembly38.fasta"; \\
echo \$FQ1; \\
echo \$FQ1; \\
export CLEANUP=1;
export REMOVE_INPUT=1; \\
echo \$RGFILE; \\
export MEM=65; \\
bsub \\
\-J "\${RGBASE}_s01alnsrt" \\
\-o "\${BASE}/WXS_Aquilla/04-LOGS/\${RGBASE}_s01alnsrt.%J" \\
\-u "\${EMAIL}" \\
\-n \${THREADS} -W 4320 \\
\-M 86000000 \\
\-R "rusage[mem=89152]" \\
\-q research-hpc \\
\-a 'docker(achalneupane/bwaref)' \\
entrypoint.sh; \\
echo -e "02 - Starting validatesam per sample \${FULLSM} and RGBASE
\${RGBASE}\\nBAMFILE:\${BAMFILE}"; \\
export MEM=16; \\
export
BAMFILE="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}/\${RGBASE}.aln.srt.bam";
\\
echo \${BAMFILE}; \\
export REF="\${BASE}/Genome_Ref/GRCh38/Homo_sapiens_assembly38.fasta"; \\
bsub \\
\-w "done(\\"\${RGBASE}_s01alnsrt\\")" \\
\-J "\${RGBASE}_s02vldate" \\
\-o "\${BASE}/WXS_Aquilla/04-LOGS/\${RGBASE}_s02vldate.%J" \\
\-u "\${EMAIL}" \\
\-n1 -W 1360 \\
\-R "rusage[mem=18192]" \\
\-q research-hpc \\
\-a "docker(achalneupane/validatesamref)" \\
entrypoint.sh -IGNORE INVALID_VERSION_NUMBER -IGNORE INVALID_TAG_NM; \\
echo -e "03 - Starting intersectbed per sample \${FULLSM} and RGBASE
\${RGBASE}\\nBAMFILE:\${BAMFILE}"; \\
export RUN_TYPE="paddedexome"; \\
export BEDFILE="\${BASE}/Genome_Ref/GRCh38/Capture_Padded.GRCh38.bed"; \\
export COVERED_BED="\${BASE}/Genome_Ref/GRCh38/Capture_Covered.GRCh38.bed"; \\
export PADDED_BED="\${BASE}/Genome_Ref/GRCh38/Capture_Padded.GRCh38.bed";\\
export REF="\${BASE}/Genome_Ref/GRCh38/Homo_sapiens_assembly38.fasta"; \\
export
BAMFILE="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}/\${RGBASE}.aln.srt.bam";
\\
bsub \\
\-w "done(\\"\${RGBASE}_s02vldate\\")" \\
\-J "\${RGBASE}_s03intsct" \\
\-o "\${BASE}/WXS_Aquilla/04-LOGS/\${RGBASE}_s03intsct.%J" \\
\-u "\${EMAIL}" \\
\-n1 -W 1360 \\
\-q research-hpc \\
\-a "docker(achalneupane/intersectbedref)" \\
entrypoint.sh; \\
echo -e "04 - Starting validatesam per sample \${FULLSM} and RGBASE
\${RGBASE}\\nBAMFILE:\${BAMFILE}"; \\
export
BAMFILE="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}/\${RGBASE}.aln.srt.isec-\${RUN_TYPE}.bam";
\\
bsub \\
\-w "done(\\"\${RGBASE}_s03intsct\\")" \\
\-J "\${RGBASE}_s04vldate" \\
\-o "\${BASE}/WXS_Aquilla/04-LOGS/\${RGBASE}_s04vldate.%J" \\
\-u "\${EMAIL}" \\
\-n1 -W 1360 \\
\-R "rusage[mem=18192]" \\
\-q research-hpc \\
\-a "docker(achalneupane/validatesamref)" \\
entrypoint.sh -IGNORE INVALID_VERSION_NUMBER -IGNORE INVALID_TAG_NM -IGNORE
MATE_NOT_FOUND; \\
echo -e "05 - Starting bamtocram per sample \${FULLSM} and RGBASE
\${RGBASE}\\nBAMFILE:\${BAMFILE}\\nOUT_DIR:\${OUT_DIR}";\\
export
BAMFILE="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}/\${RGBASE}.aln.srt.bam";
\\
export OUT_DIR="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}/"; \\
done; \\
echo -e "DONE ANALYZING RGBASES per sample \${FULLSM}";\\
IFS=\$'\\n' RGBASES=(\$(grep "\${FULLSM}" "\${LOOKUP}" \| cut -d,
\-f\${LOOKUP_COL_RGBASE})); \\
INPUT_LIST=(); \\
WAIT_LIST=(); \\
CLEANUP_LIST=(); \\
for RGBASE in \${RGBASES[@]}; do \\
INPUT_LIST+=("\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}/\${RGBASE}.aln.srt.isec-\${RUN_TYPE}.bam");
\\
WAIT_LIST+=("&&" "done(\\"\${RGBASE}_s04vldate\\")"); \\
CLEANUP_LIST+=("\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.\${FQ1EXT}");
\\
CLEANUP_LIST+=("\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.\${FQ2EXT}");
\\
done; \\
echo -e "06 - Starting markduplicates for FULLSM \${FULLSM} with
INPUT_LIST:\${INPUT_LIST[@]}"; \\
export FULLSM="\${FULLSM}"; \\
export MEM=32; \\
export OUT_DIR="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}/"; \\
bsub \\
\-w \${WAIT_LIST[@]:1} \\
\-J "\${FULLSM}_s06mrkdup" \\
\-o "\${BASE}/WXS_Aquilla/04-LOGS/\${FULLSM}_s06mrkdup.%J" \\
\-u "\${EMAIL}" \\
\-n1 -W 1440 \\
\-M 46000000 \\
\-R "rusage[mem=49152]" \\
\-q research-hpc \\
\-a "docker(achalneupane/markduplicates)" \\
\${INPUT_LIST[@]};
START=1;
END=1;
SHELLDROP=1
Snumber=1;
export BASE="/gscmnt/gc2645/wgs"; \\
export WORKDIR="\${BASE}/tmp"; \\
export THREADS=16; \\
export BWA_PIPE_SORT=1; \\
export TIMING=1; \\
LOOKUP_COL_SM=1; \\
LOOKUP_COL_DNA=2; \\
LOOKUP_COL_PR=3; \\
LOOKUP_COL_RGBASE=4; \\
LOOKUP_COL_FQEXT=5; \\
LOOKUP_COL_RGBASEtemp=6; \\
unset FBASE; \\
export RUN_TYPE="paddedexome"; \\
export FASTQ_TYPE="interleaved"; \\
export MODE="fq"; \\
for FULLSM in \$(sed -n "\${START},\${END}p" "\${WORKLIST}"); do \\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo "Doing sample number\*\*\*\*\*\*\*\*\*\*: " \${Snumber}; \\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
((Snumber=\${Snumber}+1)); \\
SM="\$(echo "\${FULLSM}" \| cut -d\^ -f1)"; \\
IFS=\$'\\n' export DNA=(\$(awk -F, "\\\$\${LOOKUP_COL_SM} == \\"\${SM}\\""
"\${LOOKUP}" \| cut -d, -f\${LOOKUP_COL_DNA} \| sort -u)); \\
if [ \${\#DNA[@]} -gt 1 ]; then echo "Warning, \\\${DNA} not unique for \${SM}
(n=\${\#DNA[@]}: \${DNA[@]})"; fi; \\
SM="\$(echo "\${FULLSM}" \| cut -d\^ -f1)"; \\
DNA="\$(echo "\${FULLSM}" \| cut -d\^ -f2)"; \\
PR="\$(echo "\${FULLSM}" \| cut -d\^ -f3)"; \\
export OUT_DIR="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}"; \\
echo -e "00 - Starting jobs per sample FULLSM \${FULLSM}"; \\
for RGBASE in \$(grep "\${SM},\${DNA},\${PR}" "\${LOOKUP}" \| cut -d,
\-f\${LOOKUP_COL_RGBASE}); do \\
FULLSMtemp="\${SM}\^unk\^\${PR}"; \\
FBASEtemp="/40/AD/AD_Seq_Data/01.-RawData/CACHE_WGS/01.-RawData/\${FULLSMtemp}";
\\
FBASE="/40/AD/AD_Seq_Data/01.-RawData/CACHE_WGS/01.-RawData/\${FULLSM}"; \\
FQEXT=(\$(awk -F, "\\\$\${LOOKUP_COL_RGBASE} == \\"\${RGBASE}\\"" "\${LOOKUP}"
\| cut -d, -f\${LOOKUP_COL_FQEXT})); \\
RGBASEtemp=(\$(awk -F, "\\\$\${LOOKUP_COL_RGBASE} == \\"\${RGBASE}\\""
"\${LOOKUP}" \| cut -d, -f\${LOOKUP_COL_RGBASEtemp})); \\
RGFILEtemp="\${FBASEtemp}/\${RGBASEtemp}.rgfile"; \\
RGFILE="\${FBASE}/\${RGBASE}.rgfile";\\
FQtemp="\${FBASEtemp}/\${RGBASEtemp}.\${FQEXT}";\\
FQ="\${FBASE}/\${RGBASE}.\${FQEXT}"; \\
DEST="\${BASE}/WXS_Aquilla/01-RAW";\\
echo -e "00a - Uploading FASTQ and rgfiles for sample \${FULLSM} and RGBASE
\${RGBASE}\\nFQ:\${FQ}\\nRGFILE:\${RGFILE}\\nDNA:\${DNA}"; \\
mkdir \${DEST}/\${PR}/\${FULLSM}; \\
rsync -avh -L \${USER}@fenix.psych.wucon.wustl.edu:\${FQtemp}
\${DEST}/\${PR}/\${FULLSM}/ ;\\
mv -f "\${DEST}/\${PR}/\${FULLSM}/\${RGBASEtemp}.\${FQEXT}"
"\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.\${FQEXT}"; \\
rsync -avh \${USER}@fenix.psych.wucon.wustl.edu:\${RGFILEtemp}
\${DEST}/\${PR}/\${FULLSM}/ ;\\
mv -f "\${DEST}/\${PR}/\${FULLSM}/\${RGBASEtemp}.rgfile"
"\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.rgfile"; \\
export RGFILE="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.rgfile";
\\
export FULLSM_RGID="\${RGBASE}"; \\
unset BAMFILE; \\
unset FQ1; \\
unset FQ2; \\
IFS=\$'\\n' FQEXT=(\$(awk -F, "\\\$\${LOOKUP_COL_RGBASE} == \\"\${RGBASE}\\""
"\${LOOKUP}" \| cut -d, -f\${LOOKUP_COL_FQEXT})); \\
echo -e "01 - Starting bwa per sample \${FULLSM} and RGBASE \${RGBASE}";\\
export RGFILE="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.rgfile";
\\
export FQ="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.\${FQEXT}";
\\
export REF="\${BASE}/Genome_Ref/GRCh38/Homo_sapiens_assembly38.fasta"; \\
export MEM=32; \\
bsub \\
\-J "\${RGBASE}_s01alnsrt" \\
\-o "\${BASE}/WXS_Aquilla/04-LOGS/\${RGBASE}_s01alnsrt.%J" \\
\-u "\${EMAIL}" \\
\-n \${THREADS} -W 2160 \\
\-M 46000000 \\
\-R "rusage[mem=49152]" \\
\-q research-hpc \\
\-a 'docker(achalneupane/bwaref)' \\
entrypoint.sh; \\
\# The rest of the code after achalneupane/bwaref is the same as in Appendix A
!!
- Helper script to feed input CRAM/BAM and unwrap them into FASTQ (replace “.crai” with “.bai” if using BAM as an input)
Snumber=1
START=1; \\
END=156; \\
DELAY=10
EMAIL="achal@wustl.edu"
SHELLDROP=0
export BASE="/gscmnt/gc2645/wgs"; \\
export WORKDIR="\${BASE}/tmp"; \\
export THREADS=16; \\
export BWA_PIPE_SORT=1; \\
export TIMING=1;\\
PR="202103_ADSP_FUS-familial_WGS_UNNAMED"; \\
WORKLIST="\${BASE}/WXS_Aquilla/01-RAW/\${PR}-worklist_fullsm.csv"; \\
LOOKUP="\${BASE}/WXS_Aquilla/01-RAW/\${PR}-MGIID-sm-dna-pr-rgbase-cram-smdir-cramloc.csv";
\\
LOOKUP_COL_SM=2; \\
LOOKUP_COL_DNA=3; \\
LOOKUP_COL_PR=4; \\
LOOKUP_COL_CRAMFILE=5; \\
export RUN_TYPE="paddedexome"; \\
for FULLSM in \$(sed -n "\${START},\${END}p" "\${WORKLIST}"); do \\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo "Doing sample
number\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*:
" \$Snumber; \\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
echo
"\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*\*";
\\
((Snumber=Snumber+1)); \\
export FULLSM="\${FULLSM}"; \\
SM="\$(echo "\${FULLSM}" \| cut -d\^ -f1)"; \\
IFS=\$'\\n' export DNA=(\$(awk -F, "\\\$\${LOOKUP_COL_SM} == \\"\${SM}\\""
"\${LOOKUP}" \| cut -d, -f\${LOOKUP_COL_DNA} \| sort -u)); \\
if [ \${\#DNA[@]} -gt 1 ]; then echo "Warning, \\\${DNA} not unique for
\${SM} (n=\${\#DNA[@]}: \${DNA[@]})"; fi; \\
DNA="\$(echo "\${FULLSM}" \| cut -d\^ -f2)"; \\
PR="\$(echo "\${FULLSM}" \| cut -d\^ -f3)"; \\
CRAM="\$(grep "\${SM},\${DNA},\${PR}" "\${LOOKUP}" \| cut -d,
\-f\${LOOKUP_COL_CRAMFILE})"
export OUT_DIR="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}"; \\
INPUT_LIST=(); \\
WAIT_LIST=(); \\
rsync -avh \${USER}@fenix.psych.wucon.wustl.edu:\${CRAMLOC}/\${CRAM}
\${OUT_DIR}/; \\
rsync -avh \${USER}@fenix.psych.wucon.wustl.edu:\${CRAMLOC}.\${CRAM}.crai
\${OUT_DIR}/; \\
export MEM=16; \\
export CREATE_RGFILE=1; \\
export DEBUG=1; \\
export REMOVE_INPUT=1; \\
export OUT_DIR="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}" ;\\
export REF="\${BASE}/Genome_Ref/GRCh38/Homo_sapiens_assembly38.fasta"; \\
export CRAMBASE="\$(echo \${CRAM/.cram/})"; \\
export CRAMFILE="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${CRAM}"; \\
echo -e "OUTPUT is in
"\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${CRAMBASE}_s00rvtcram.%J"
\\n OUTDIR is \${OUT_DIR}";\\
bsub \\
\-J "\${FULLSM}_s00rvtcram" \\
\-o
"\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${CRAMBASE}_s00rvtcram.%J"
\\
\-u "\${EMAIL}" \\
\-n1 -W 1440 \\
\-R "rusage[mem=18192]" \\
\-q research-hpc \\
\-a 'docker(achalneupane/cram2fastq)' \\
/bin/bash; \\
echo -e "00b - Starting revertcram for
\${FULLSM}\\n\${CRAMFILE}\\n\${SM}\\n\${DNA}\\n\${PR}"; \\
SAMTOOLS="/gscmnt/gc2645/wgs/variant_calling/samtools"; \\
IFS=\$'\\n' RGS=(\$(\${SAMTOOLS} view -H "\${CRAMFILE}" \| grep "\^@RG"));
\\
for RG in \${RGS[@]}; do RGID="\$(echo \${RG} \| grep -oP
"(?\<=ID:)[\^[:space:]]\*")"; \\
RGID_NEW="\$(echo \${RGID} \| cut -d: -f2- \| sed 's/:/\^/g')"; \\
RGBASE="\${FULLSM}.\${RGID_NEW}"; \\
echo -e "01 - Starting bwa per sample \${FULLSM} and RGBASE
\${RGBASE}\\nFQ1:\${FQ1}\\nFQ2:\${FQ2}\\nRGFILE:\${RGFILE}\\nDNA:\${DNA}\\nPR:\${PR}\\nIFS:\${IFS}";\\
export
RGFILE="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.rgfile"; \\
export FULLSM_RGID="\${RGBASE}"; \\
unset BAMFILE; \\
export OUT_DIR="\${BASE}/WXS_Aquilla/02-TRANSIT/\${PR}/\${FULLSM}"; \\
FQ1EXT="r1.fastq"; \\
FQ2EXT="r2.fastq"; \\
export
FQ1="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.\${FQ1EXT}";
\\
export
FQ2="\${BASE}/WXS_Aquilla/01-RAW/\${PR}/\${FULLSM}/\${RGBASE}.\${FQ2EXT}";
\\
export REF="\${BASE}/Genome_Ref/GRCh38/Homo_sapiens_assembly38.fasta"; \\
export MEM=52; \\
export CLEANUP=1;
export REMOVE_INPUT=1; \\
bsub \\
\-w "done(\\"\${FULLSM}_s00rvtcram\\")" \\
\-J "\${RGBASE}_s01alnsrt" \\
\-o "\${BASE}/WXS_Aquilla/04-LOGS/\${RGBASE}_s01alnsrt.%J" \\
\-u "\${EMAIL}" \\
\-n \${THREADS} -W 2880 \\
\-M 66000000 \\
\-R "rusage[mem=69152]" \\
\-q research-hpc \\
\-a 'docker(achalneupane/bwaref)' \\
/bin/bash; \\
- The rest of the code after achalneupane/bwaref is the same as in Appendix A
- Docker image for achalneupane/cram2fastq
\# RevertSam and SamToFastq are the tools that unwraps the CRAM or BAM files
into paired FASTQ per RG ID
CUR_STEP="RevertSam"
start=\$(\${DATE}); echo "[\$(display_date \${start})] \${CUR_STEP}
starting"
"\${TIMING[@]}" /usr/local/openjdk-8/bin/java \${JAVAOPTS} -jar "\${PICARD}"
\\
"\${CUR_STEP}" \\
\-I "\${CRAMFILE}" \\
\-R \${REF} \\
\-O /dev/stdout \\
\-SORT_ORDER queryname \\
\-COMPRESSION_LEVEL 0 \\
\-VALIDATION_STRINGENCY SILENT \\
\| /usr/local/openjdk-8/bin/java \${JAVAOPTS} -jar "\${PICARD}" \\
SamToFastq \\
\-I /dev/stdin \\
\-R \${REF} \\
\-OUTPUT_PER_RG true \\
\-RG_TAG ID \\
\-OUTPUT_DIR "\${OUT_DIR}" \\
\-VALIDATION_STRINGENCY SILENT
\# Transfer of files
CUR_STEP="Transfer of files"
start=\$(\${DATE}); echo "[\$(display_date \${start})] \${CUR_STEP}
starting"
CRAMLINES=\$(samtools idxstats "\${CRAMFILE}" \| awk '{s+=\$3+\$4} END
{print s\*4}')
FQLINES=\$(cat \${OUT_DIR}/\*.fastq \| wc -l)
if [ ! -z \${DEBUG} ]; then
echo "\${CRAMLINES} lines in .cram/bam"
echo "\${FQLINES} lines in all .fastq files"
if [ \$(echo "scale=2;\${FQLINES}/\${CRAMLINES} \> 0.90" \| bc) -eq 0 ];
then
echo "Warning, .fastq files contain less than 90% of the number of reads of
.cram/bam file"
fi
fi
\#\# Create RGFILE
CUR_STEP="Create RGFILE"
start=\$(\${DATE}); echo "[\$(display_date \${start})] \${CUR_STEP}
starting"
if [ ! -z "\${CREATE_RGFILE}" ]; then
SM="\$(echo "\${FULLSM}" \| cut -d\^ -f1)"
DNA="\$(echo "\${FULLSM}" \| cut -d\^ -f2)"
PR="\$(echo "\${FULLSM}" \| cut -d\^ -f3)"
IFS=\$'\\n' RGS=(\$(samtools view -H "\${CRAMFILE}" \| grep "\^@RG"))
echo "Creating \${\#RGS[@]} .rgfiles for newly created .fastq files"
echo "Moving \${\#RGS[@]} .fastq files to \${OUT_DIR}/"
for RG in \${RGS[@]}; do
RGID="\$(echo \${RG} \| grep -oP "(?\<=ID:)[\^[:space:]]\*")"
RGID_NEW="\$(echo \${RGID} \| cut -d: -f2- \| sed 's/:/\^/g')"
mv -vf "\${OUT_DIR}/\${RGID//:/_}_1.fastq"
"\${OUT_DIR}/\${FULLSM}.\${RGID_NEW}.r1.fastq"
if [ -f "\${OUT_DIR}/\${RGID//:/_}_2.fastq" ]; then mv -vf
"\${OUT_DIR}/\${RGID//:/_}_2.fastq"
"\${OUT_DIR}/\${FULLSM}.\${RGID_NEW}.r2.fastq"; fi
RGPU="\$(echo \${RG} \| grep -oP "(?\<=PU:)[\^[:space:]]\*")"
RGLB="\${SM}.\${PR}"
echo
"@RG\\tID:\${RGID}\\tPL:illumina\\tPU:\${RGPU}\\tLB:\${RGLB}\\tSM:\${SM}\\tDS:\${SM}\^\${DNA}\^\${PR}"
\> "\${OUT_DIR}/\${FULLSM}.\${RGID_NEW}.rgfile"
done
fi
\#\# Cleaning up files
CUR_STEP="Cleaning up files"
start=\$(\${DATE}); echo "[\$(display_date \${start})] \${CUR_STEP}
starting"
if [ \${exitcode} -eq 0 ] && [ \${REMOVE_INPUT} -eq 1 ]; then
rm -fv "\${CRAMFILE}" "\${CRAMFILE%.cram}.crai" "\${CRAMFILE}.crai"
2\>/dev/null
echo -e "REMOVE_INPUT was set to \${REMOVE_INPUT} then CRAMFILE is removed"
else
echo -e "REMOVE_INPUT was set to \${REMOVE_INPUT} then CRAMFILE is NOT
removed"
fi
- Docker image (achalneupane/bam2fqv2) to extract interleaved FASTQ from BAM
\# Extract read Group information
IFS=\$'\\n' RGS=(\$(samtools view -@ \${THREADS} -h \${BAMFILE} \| head -n
10000000 \| grep \^HS2000 \| cut -d\$'\\t' -f1\| cut -d: -f1,2 \| sort -V \|
uniq \| grep \^HS2000))
echo "Readgroups are \${RGS[@]}"
unset IFS
args=(tee)
for RG in \${RGS[@]}; do
args+=(\\\>\\(grep -A3 --no-group-separator \\"\^@\${RG/\^/:}:\\" \\\| gzip
\\\> \${OUT_DIR}/\${SM}\^\${DNA}\^\${PR}.\${RG/:/.}.fq.gz\\))
done
args+=(\\\>/dev/null)
JAVA="/usr/local/openjdk-8/bin/java"
\# JAVAOPTS="-Xms2g -Xmx\${MEM}g -XX:+UseSerialGC
\-Dpicard.useLegacyParser=false"
JAVAOPTS="-Xms4g -Xmx\${MEM}g -XX:ParallelGCThreads=\${THREADS}
\-Djava.io.tmpdir=\${TMP_DIR}"
CUR_STEP="RevertSam"
start=\$(\${DATE}); echo "[\$(display_date \${start})] \${CUR_STEP}
starting"
"\${TIMING[@]}" \${JAVA} \${JAVAOPTS} -jar "\${PICARD}" \\
"\${CUR_STEP}" \\
I="\${BAMFILE}" \\
O=/dev/stdout \\
SORT_ORDER=queryname \\
COMPRESSION_LEVEL=0 \\
VALIDATION_STRINGENCY=SILENT \\
TMP_DIR=\${TMP_DIR} \\
\| \${JAVA} \${JAVAOPTS} -jar "\${PICARD}" \\
SamToFastq \\
I=/dev/stdin \\
FASTQ=/dev/stdout \\
INTERLEAVE=TRUE \\
VALIDATION_STRINGENCY=SILENT \\
TMP_DIR=\${TMP_DIR} \| eval \${args[@]}
arr=(\${PIPESTATUS[@]}); exitcode=0; for i in \${arr[@]}; do
((exitcode+=i)); done
- The rest of the pipeline is the same as in Appendix C + Appendix B
- Function to download SRA and extract FASTQ
getSRAtoFastq()
{
DIR=\$1
SAMPLE=\$2
mkdir -p \${DIR}/\${SAMPLE}
SRR="\$(echo \$SAMPLE \| cut -d\^ -f2)"
echo "Doing: " \${SRR}
\# LOOKUP="/30/dbGaP/6109/sra/lookup.csv"
\# If \*.Confirm.txt file is not present; then only run this. We will also
validate SRA and fastqs as we download/process.
if ! [ -f \${DIR}/\${SAMPLE}/\*".Confirm.txt" ]; then
prefetch --ngc /30/dbGaP/6109/prj_6109.ngc \${SRR}
vdb-validate ./\${SRR} 2\> \>(grep -i Column)
vdb-validate ./\${SRR} 2\> "\${DIR}/vdbValidate/vdb-validate_\${SRR}.txt"
cat "\${DIR}/vdbValidate/vdb-validate_\${SRR}.txt" \| grep Column \>
"\${DIR}/vdbValidate/Columns_\${SRR}.txt"
col_numbers="\$(cat "\${DIR}/vdbValidate/Columns_\${SRR}.txt" \| wc -l)"
ok_numbers="\$(cat "\${DIR}/vdbValidate/Columns_\${SRR}.txt" \| grep ok \|
wc -l)"
if [ \${col_numbers} -eq \${ok_numbers} ] && [ \${col_numbers} -ne 0 ]; then
\#start fq split
echo \$col_numbers "Cols are equal " \$ok_numbers "OKs" \>
"\${DIR}/\${SAMPLE}/\${SAMPLE}.txt"
IFS=\$'\\n'
RGLINES=(\$(sam-dump --ngc /30/dbGaP/6109/prj_6109.ngc ./\${SRR} \| sed -n
'/\^[\^@]/!p;//q' \| grep \^@RG))
args=(tee)
for RGLINE in \${RGLINES[@]}; do
unset IFS
RG=(\${RGLINE})
args+=(\\\>\\(grep -A3 --no-group-separator \\"\\\\.\${RG[1]\#ID:}/[12]\$\\"
\\\| gzip \\\>
"./\${SRR}.\${RG[1]\#ID:}.fastq-dump.split.defline.z.tee.fq.gz"\\))
done
args+=(\\\>/dev/null)
echo "Splitting \${SRR}.sra into \${\#RGLINES[@]} ReadGroups"
\#\#\# NOTE: split-e wouldn't work in the downstream pipeline!!!!!
\# wget
http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
\# fastq-dump --ngc /30/dbGaP/6109/prj_6109.ngc --skip-technical --split-e
\--defline-seq '@\$ac.\$si.\$sg/\$ri' --defline-qual '+' -Z "./\${SRR}" \|
eval \${args[@]}
/30/dbGaP/6109/sra/phs000572_201802/fqgz/test/sratoolkit.2.10.8-ubuntu64/bin/fastq-dump-orig.2.10.8
\--ngc /30/dbGaP/6109/prj_6109.ngc --split-3 --defline-seq
'@\$ac.\$si.\$sg/\$ri' --defline-qual '+' -Z "./\${SRR}" \| eval \${args[@]}
if [ \$? -ne 0 ]; then
echo "Error running fastq-dump, exiting."
exit 1
fi
\# Validate the .fq.gz that are produced. Ensuring the dowloand was not
faulty
if [ \$(ls "./\${SRR}"\*fq 2\>/dev/null \| wc -l) -eq 0 ]; then
if [ \$(ls "./\${SRR}"\*fq.gz 2\>/dev/null \| wc -l) -eq 0 ]; then
echo "Error, cannot find any .fq or .fq.gz files for \${SRR}"
exit 1
else
MODE=gz
EXT="fq.gz"
fi
else
MODE=fq
EXT="fq"
fi
echo "Validating .\${EXT} created from \${SRR}"
exitcode=0
IFS=\$'\\n'
SRAINFO=(\$(/usr/local/genome/bin/sra-stat --ngc /30/dbGaP/6109/prj_6109.ngc
\--quick ./\${SRR}))
for line in \${SRAINFO[@]}; do
IFS="\|"
split1=(\${line})
RG=\${split1[1]}
IFS=":"
split2=(\${split1[2]})
READS=\${split2[0]}
((READS\*=8))
unset IFS
echo -n "Checking \${SRR} ReadGroup \${RG}, expect \${READS} lines..."
if [ \${MODE} = "gz" ]; then
LINES=\$(zcat "./\${SRR}.\${RG}.fastq-dump.split.defline.z.tee.\${EXT}" \|
wc -l)
elif [ \${MODE} = "fq" ]; then
LINES=\$(wc -l "./\${SRR}.\${RG}.fastq-dump.split.defline.z.tee.\${EXT}")
fi
echo "found \${LINES}"
if [ \${READS} -ne \${LINES} ]; then
((exitcode+=1))
fi
done
if [ \${exitcode} -eq 0 ]; then
SM="\$(echo \$SAMPLE \| cut -d\^ -f1)";
DNA="\${SRR}"
PR="\$(echo \$SAMPLE \| cut -d\^ -f3)";
FULLSM="\${SM}\^\${DNA}\^\${PR}"
IFS=\$'\\n'
for RGLINE in \${RGLINES[@]}; do
OLD_RGID=\$(echo \${RGLINE} \| grep -o "ID:[\^[:space:]]\*" \| sed
's/ID://g')
NEW_RGID=\$(echo \${OLD_RGID} \| sed 's/\\./\^/g;s/_/\^/g')
mv "\${SRR}.\${OLD_RGID}.fastq-dump.split.defline.z.tee.\${EXT}"
"./\${FULLSM}/\${FULLSM}.\${NEW_RGID}.\${EXT}"
echo \${RGLINE} \| sed
"s@\\bSM:\\([\^[:space:]]\*\\)\\([[:space:]]\\)@SM:\${SM}\\2@g;s/\\t/\\\\\\t/g"
\> "./\${FULLSM}/\${FULLSM}.\${NEW_RGID}.rgfile"
done
echo "All spots from \${SRR} are represented in associated .\${EXT} files"
\> "./\${FULLSM}/\${FULLSM}.Confirm.txt"
rm -rf \${SRR}
unset IFS
elif [ \${exitcode} -ne 0 ]; then
echo "Errors encountered" \>\> "\${DIR}/DIRerrors/\${SAMPLE}.txt"
rm -rf \${SRR}
fi
fi
rm -rf \${SRR}
srr_count="\$(find \${DIR}/\*/\*.Confirm.txt \| wc -l)";
echo "Now getting SRR count: " \$srr_count
fi
}
- Run the function getSRAtoFastq
export -f getSRAtoFastq export \${DIR} export
WORKLIST="\${DIR}/phs000572_201707_94samples_achal.csv" export srr_count=0
\# I will download 8 SRAs in parallel
while [ \${srr_count} -lt 94 ]; do parallel -j8 getSRAtoFastq \${DIR} {}
:::: "\${WORKLIST}"
srr_count="\$(find \${DIR}/\*/\*.Confirm.txt \| wc -l)"; echo "Now getting
SRR count: " \$srr_count done
- The rest of the pipeline is the same as in Appendix B
1. GenomicsDBIMPORT
EMAIL="achal@wustl.edu"
arr="\$(echo {1..22} X Y)"
len=\${\#arr[\*]}
\#iterate with a for loop
export BASE="/gscmnt/gc2645/wgs"; \\
export TILEDB_DISABLE_FILE_LOCKING=1; \\
export MEM=40; \\
export mylist="\${BASE}/WXS_Aquilla/03-FINAL/VCFs/ID_LIST_genomicsDB.list";
\\
\# /tmp folder on MGI is limited to 250gb, but this process requires \~3TB
of tmp space to process \~8000 samples
export tmpPATH="\${BASE}/WXS_Aquilla/gvcfTest/temp/"; \\
export THREADS=16;
for (( i=0; i\<len; i++ ));
do
CHR="chr\${arr[\$i]}"
echo "Doing ::" \${CHR}
\# Define WORKING Directory
export WORKDIR="\${BASE}/WXS_Aquilla//03-FINAL/VCFs/\${CHR}"; \\
bsub \\
\-J "OtoDB\${CHR}" \\
\-u "\${EMAIL}" \\
\-n \${THREADS} -W 25160 \\
\-M 490000000 \\
\-R "rusage[mem=49152]" \\
\-o "\${BASE}/WXS_Aquilla//03-FINAL/VCFs/\${CHR}.%J" \\
\-q research-hpc \\
\-a "docker(broadinstitute/gatk:4.1.2.0)" \\
/gatk/gatk --java-options "-Xms4G -Xmx\${MEM}G
\-DGATK_STACKTRACE_ON_USER_EXCEPTION=true" GenomicsDBImport \\
\--genomicsdb-workspace-path \${WORKDIR} \\
\--batch-size 50 \\
\-L \${CHR} \\
\--sample-name-map \${mylist} \\
\--tmp-dir=\${tmpPATH} \\
\--max-num-intervals-to-import-in-parallel 10 \\
\--reader-threads \${THREADS}
Done
- GenotypeGVCFs
EMAIL="achal@wustl.edu"
arr="\$(echo {1..22} X Y)"
len=\${\#arr[\*]}
\#iterate with for loop
cd /gscmnt/gc2645/wgs/WXS_Aquilla/03-FINAL/VCFs/
export BASE="/gscmnt/gc2645/wgs"; \\
export TILEDB_DISABLE_FILE_LOCKING=1; \\
export REF="\${BASE}/Genome_Ref/GRCh38/Homo_sapiens_assembly38.fasta"; \\
export tmpPATH="--tmp-dir=\${BASE}/WXS_Aquilla/gvcfTest/temp"; \\
for (( i=0; i\<len; i++ )); \\
do
CHR="chr\${arr[\$i]}"; \\
echo "Doing :: \${CHR}"; \\
\# Make sure you have three ///
export DB="-V gendb:///\${BASE}/WXS_Aquilla/03-FINAL/VCFs/\${CHR}"; \\
export
OUTvcf="\${BASE}/WXS_Aquilla/03-FINAL/VCFs/Bloomfield_\${CHR}.vcf.gz"; \\
export MEM=40; \\
bsub \\
\-J "Geno\${CHR}" \\
\-u "\${EMAIL}" \\
\-n1 -W 25160 \\
\-M 49000000 \\
\-R "rusage[mem=49152]" \\
\-o
"\${BASE}/WXS_Aquilla/03-FINAL/VCFs/Bloomfield_joint_call_logs_\${CHR}.%J"
\\
\-q research-hpc \\
\-a "docker(broadinstitute/gatk:4.1.2.0)" \\
/gatk/gatk --java-options "-Xms4G -Xmx\${MEM}G
\-DGATK_STACKTRACE_ON_USER_EXCEPTION=true" GenotypeGVCFs \\
\-R \${REF} \\
\${DB} \\
\-O \${OUTvcf} \\
\${tmpPATH}
sleep 20
done